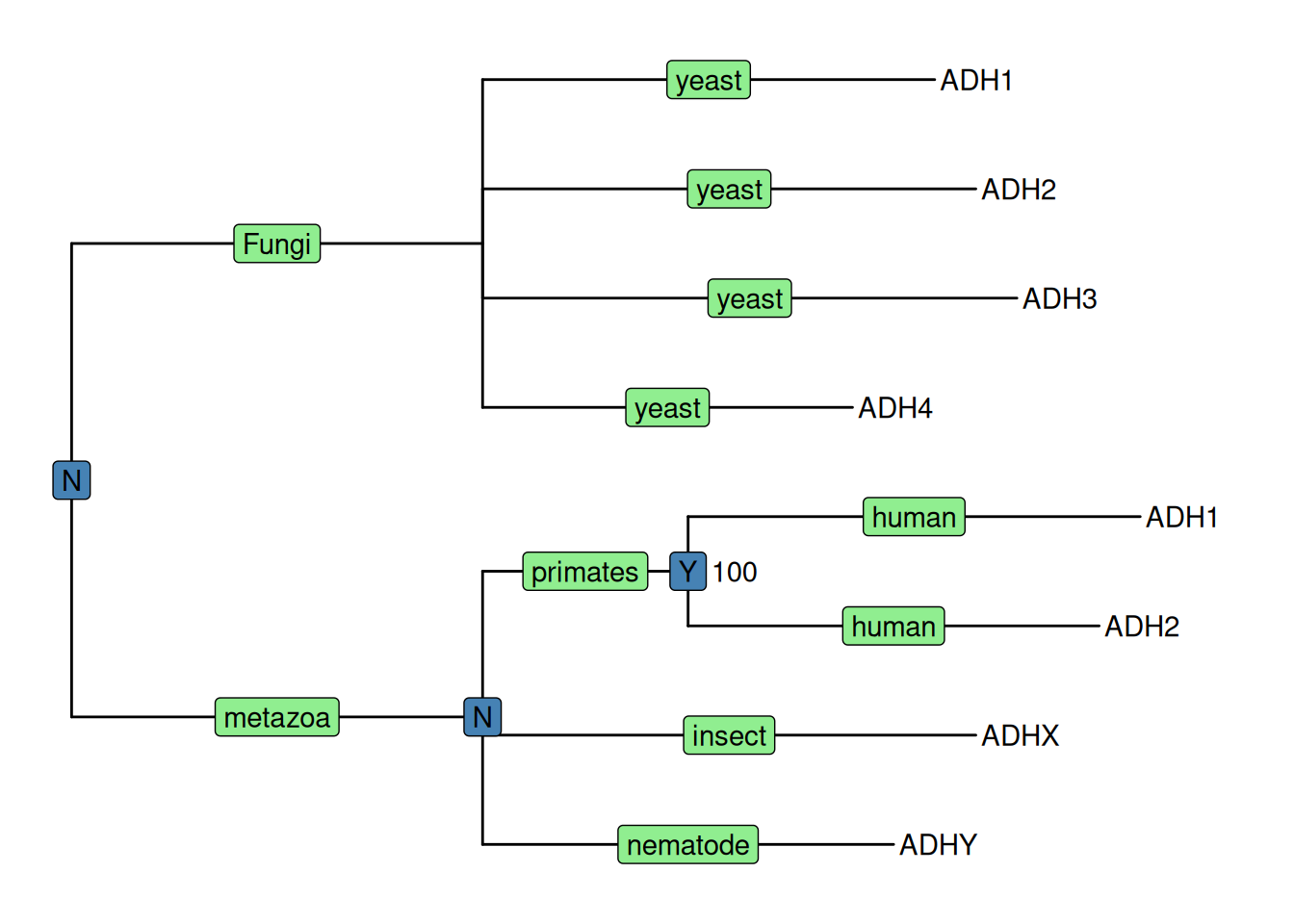
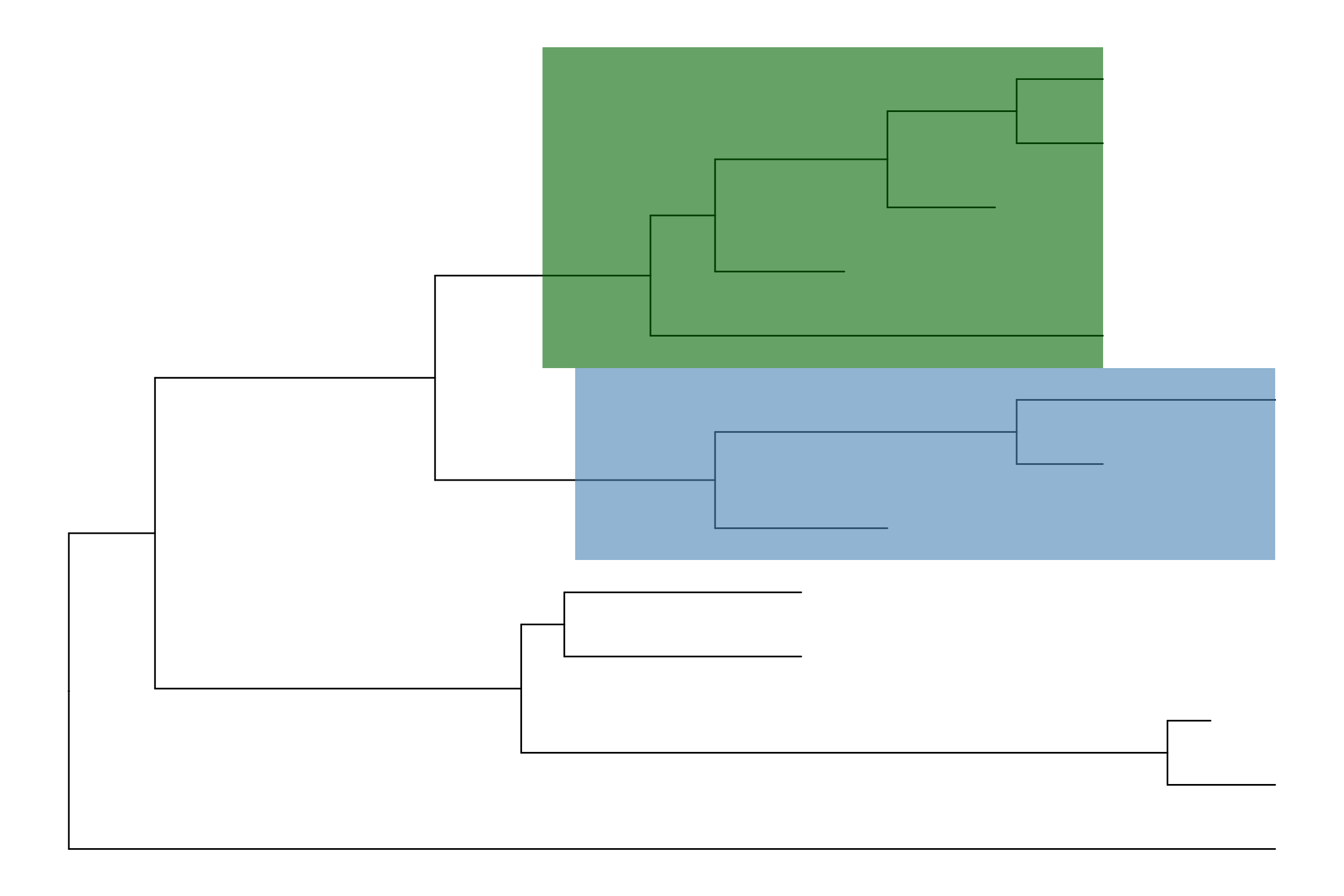
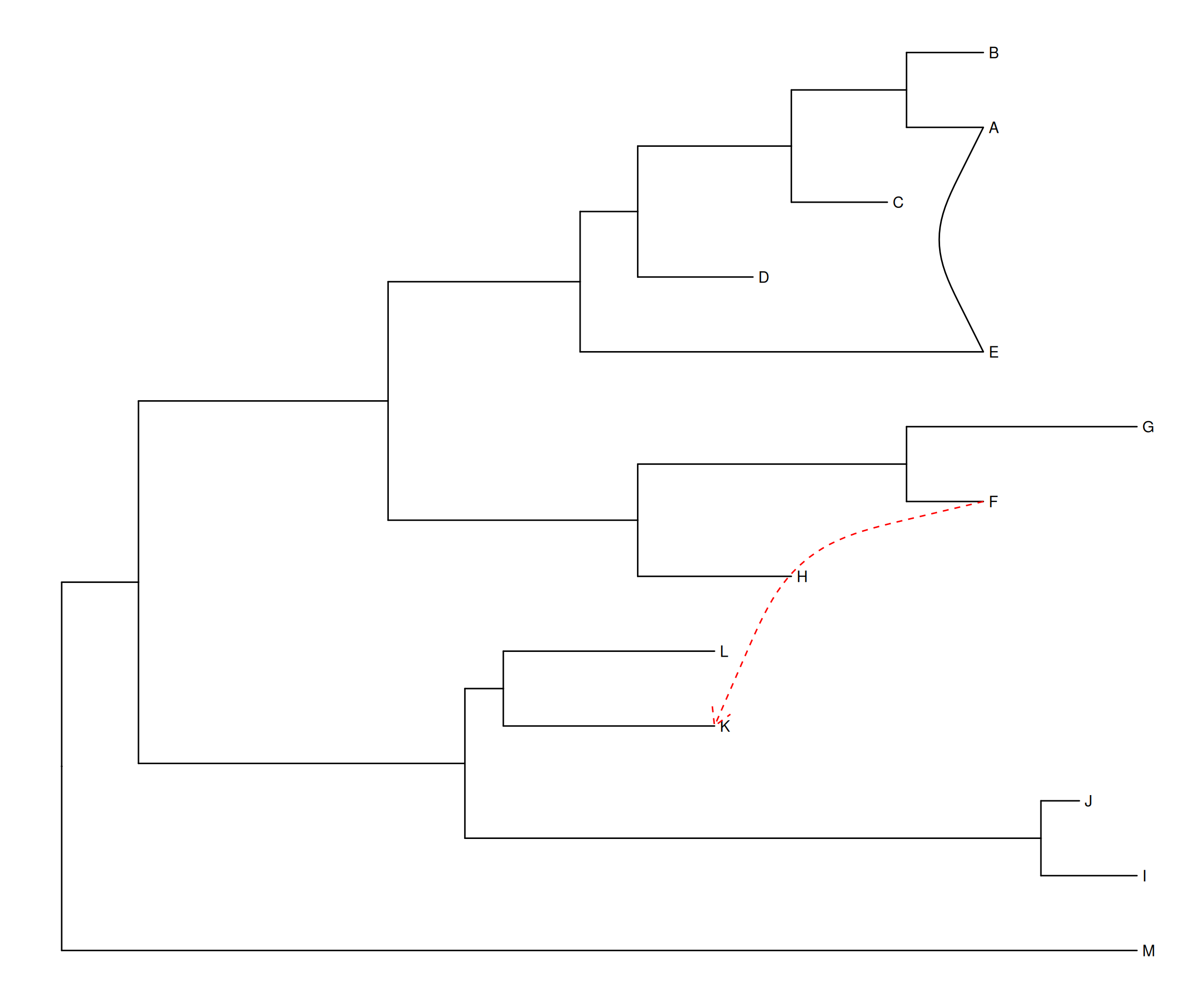
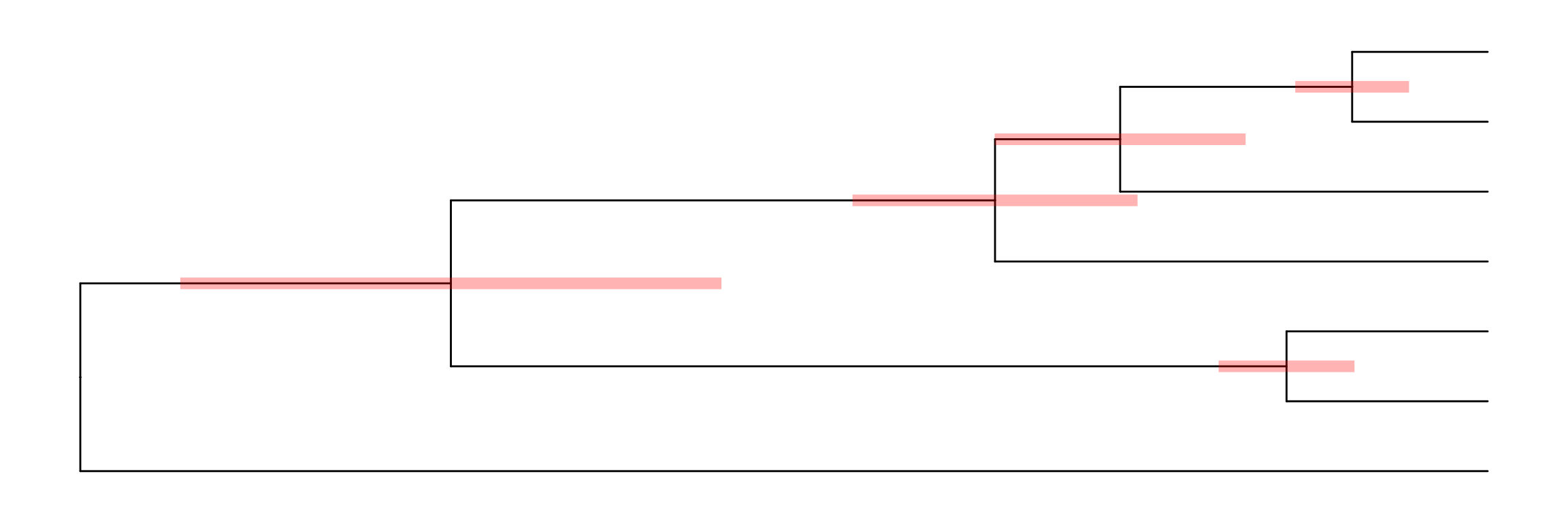
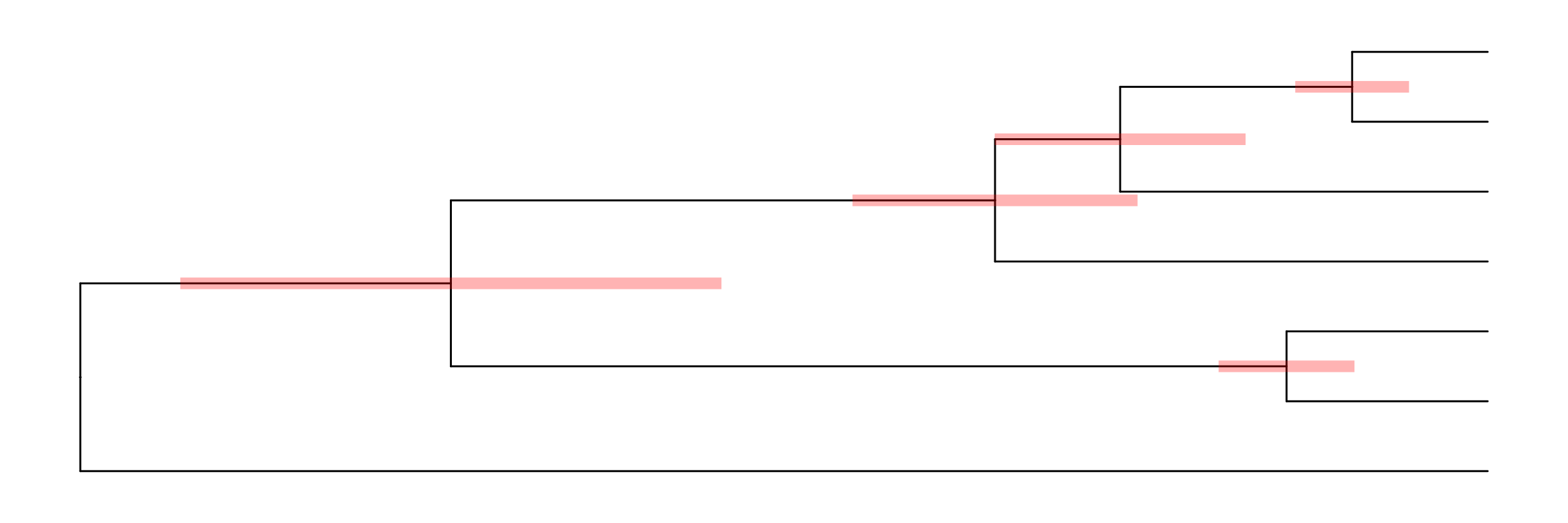
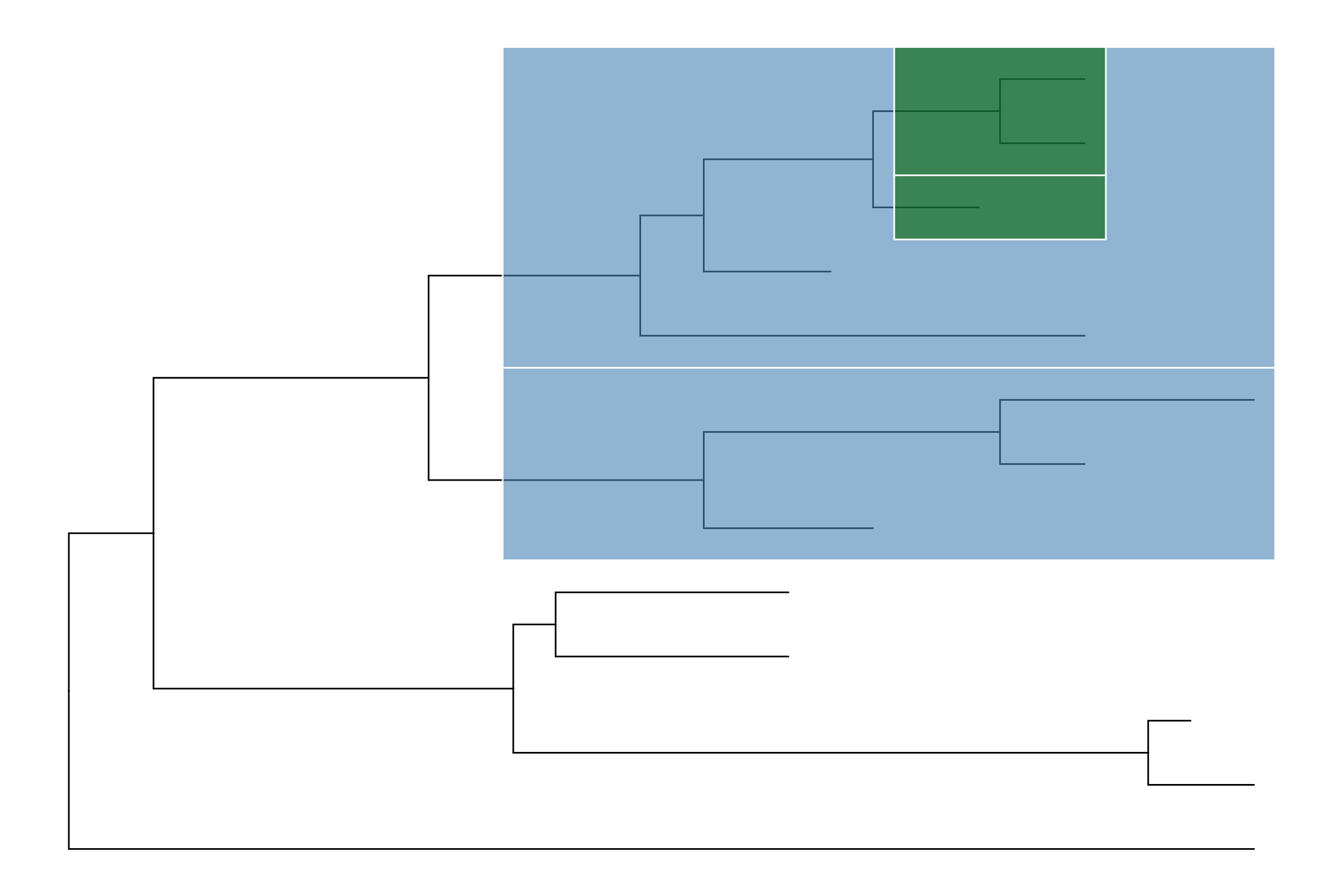p1 <- ggtree(tree) + geom_tiplab() + geom_taxalink(taxa1='A', taxa2='E') +
geom_taxalink(taxa1='F', taxa2='K', color='red', linetype = 'dashed',
arrow=arrow(length=unit(0.02, "npc")))
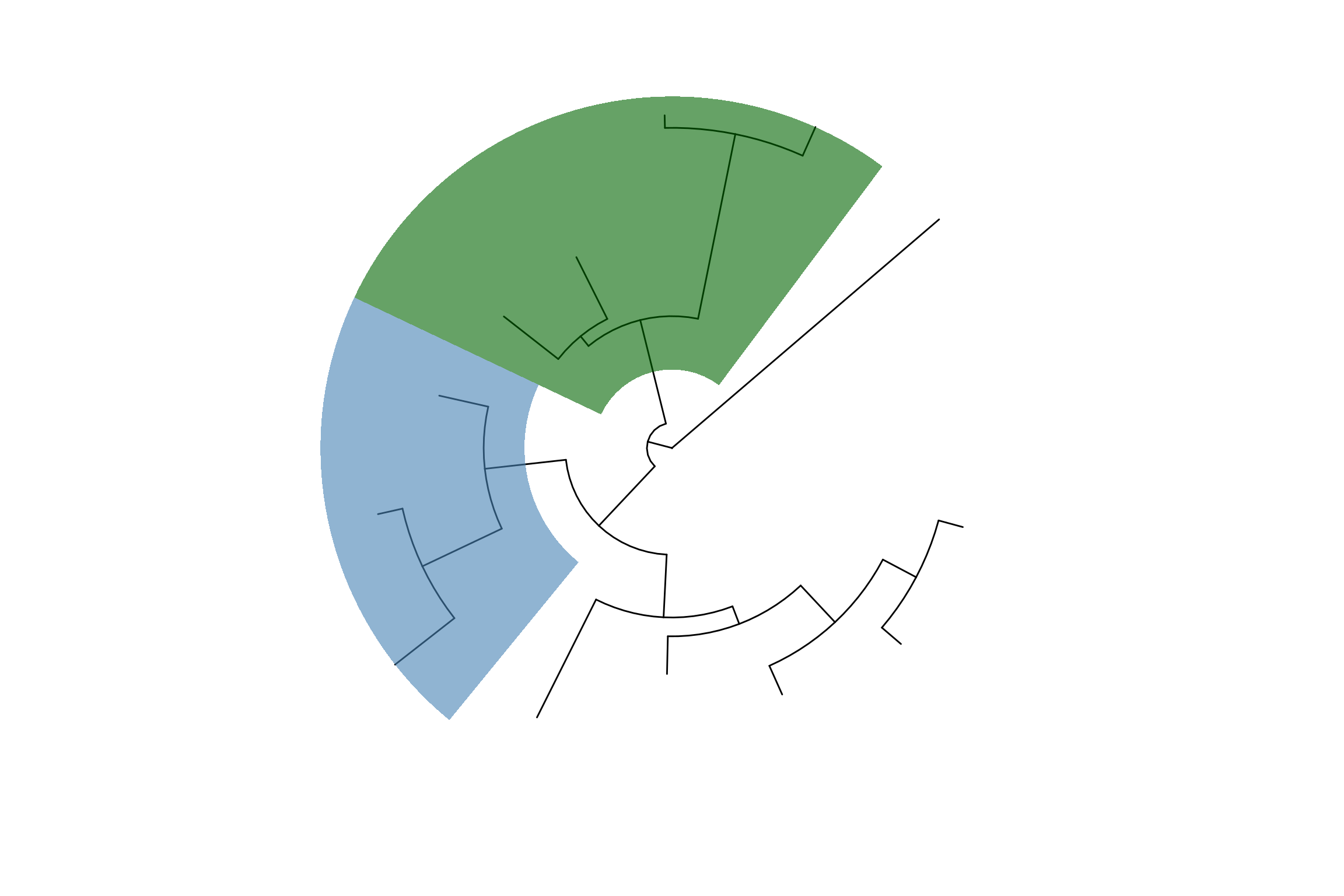
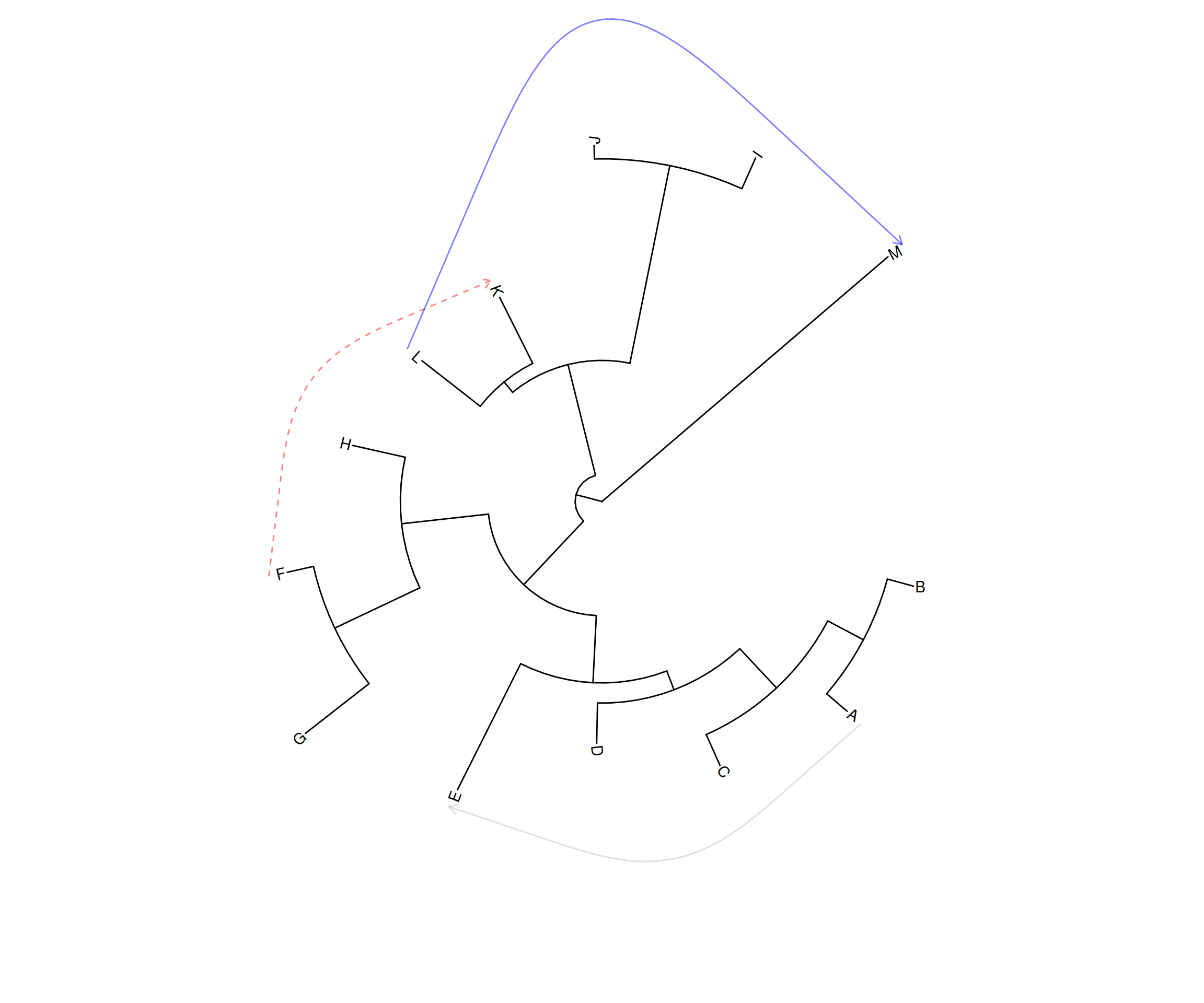
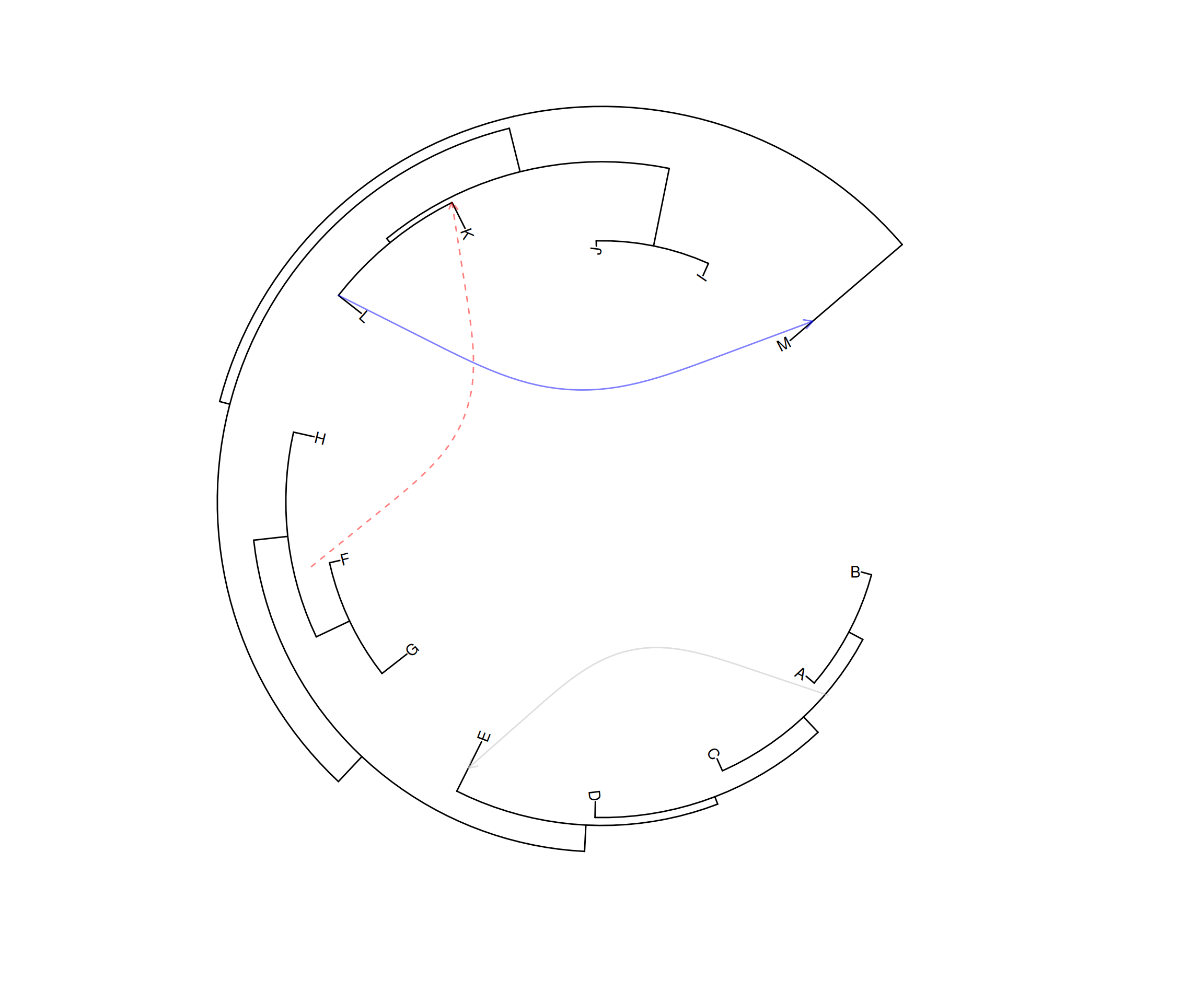
p2 <- ggtree(tree, layout="circular") +
geom_taxalink(taxa1='A', taxa2='E', color="grey", alpha=0.5,
offset=0.05, arrow=arrow(length=unit(0.01, "npc"))) +
geom_taxalink(taxa1='F', taxa2='K', color='red',
linetype = 'dashed', alpha=0.5, offset=0.05,
arrow=arrow(length=unit(0.01, "npc"))) +
geom_taxalink(taxa1="L", taxa2="M", color="blue", alpha=0.5,
offset=0.05, hratio=0.8,
arrow=arrow(length=unit(0.01, "npc"))) +
geom_tiplab()
# when the tree was created using reverse x,
# we can set outward to FALSE, which will generate the inward curve lines.
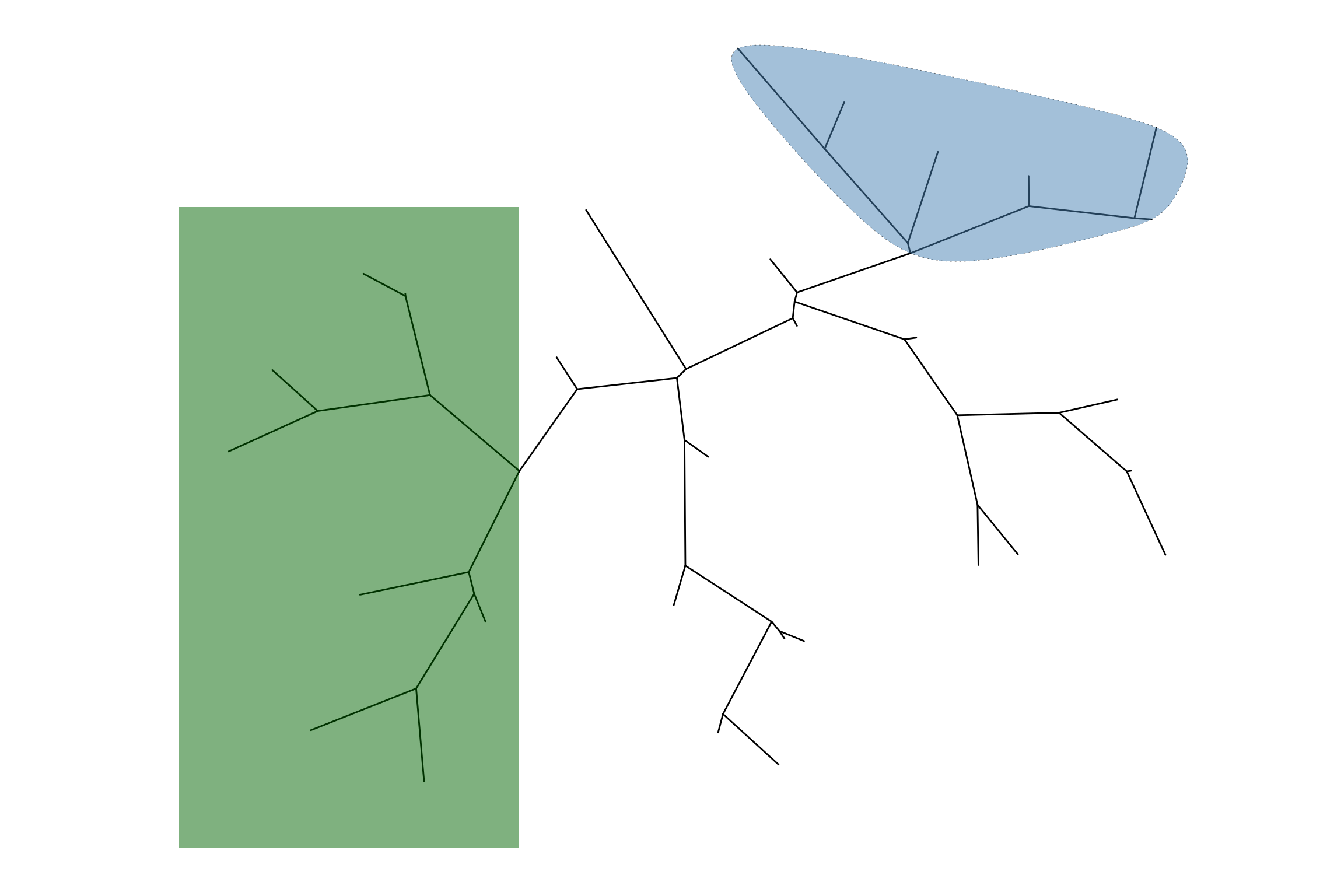
p3 <- ggtree(tree, layout="inward_circular", xlim=150) +
geom_taxalink(taxa1='A', taxa2='E', color="grey", alpha=0.5,
offset=-0.2, outward=FALSE,
arrow=arrow(length=unit(0.01, "npc"))) +
geom_taxalink(taxa1='F', taxa2='K', color='red', linetype = 'dashed',
alpha=0.5, offset=-0.2, outward=FALSE,
arrow=arrow(length=unit(0.01, "npc"))) +
geom_taxalink(taxa1="L", taxa2="M", color="blue", alpha=0.5,
offset=-0.2, outward=FALSE,
arrow=arrow(length=unit(0.01, "npc"))) +
geom_tiplab(hjust=1)
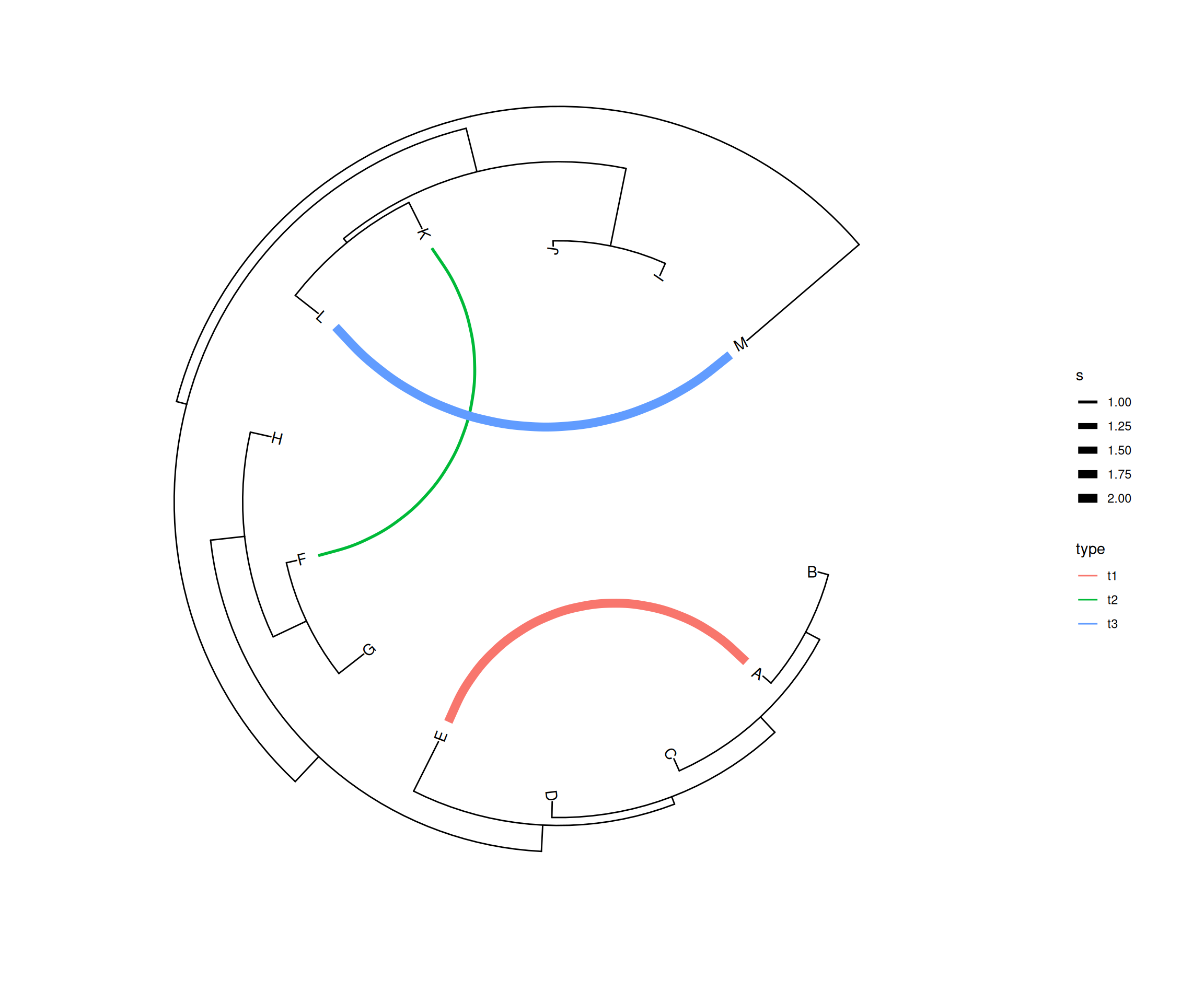
dat <- data.frame(from=c("A", "F", "L"),
to=c("E", "K", "M"),
h=c(1, 1, 0.1),
type=c("t1", "t2", "t3"),
s=c(2, 1, 2))
p4 <- ggtree(tree, layout="inward_circular", xlim=c(150, 0)) +
geom_taxalink(data=dat,
mapping=aes(taxa1=from,
taxa2=to,
color=type,
size=s),
ncp=10,
offset=0.15) +
geom_tiplab(hjust=1) +
scale_size_continuous(range=c(1,3))
p1
p2
p3
p4