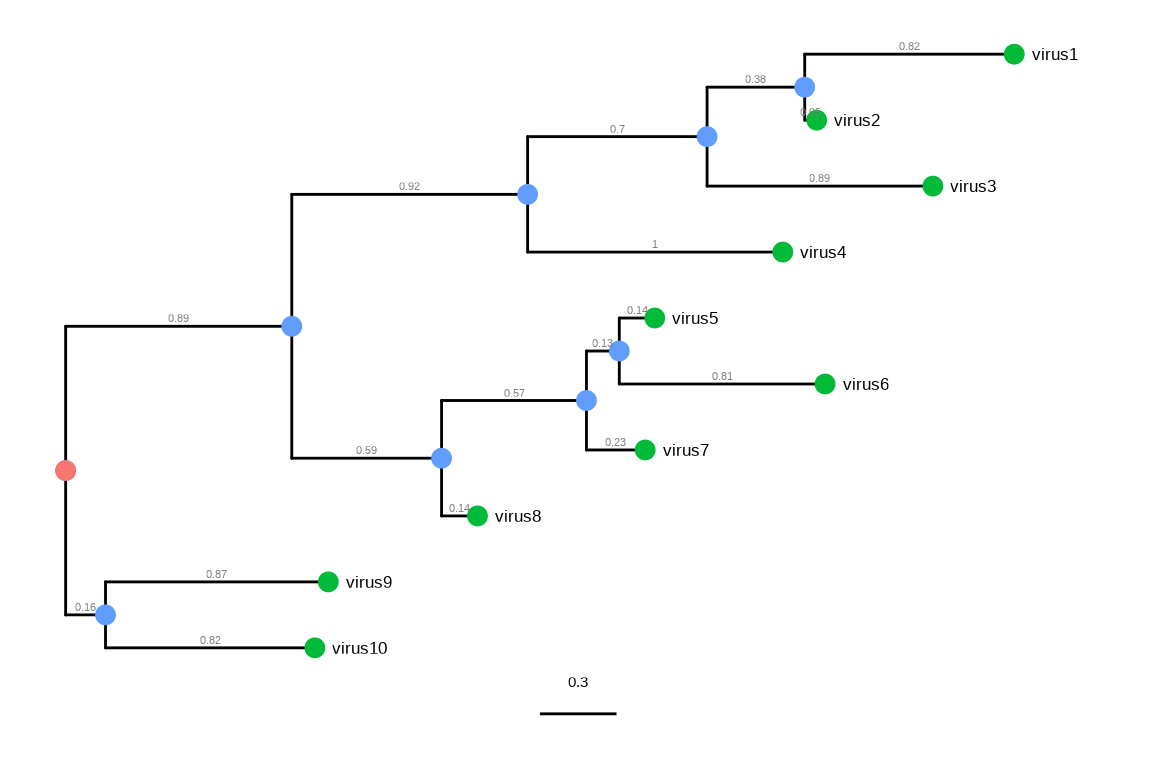
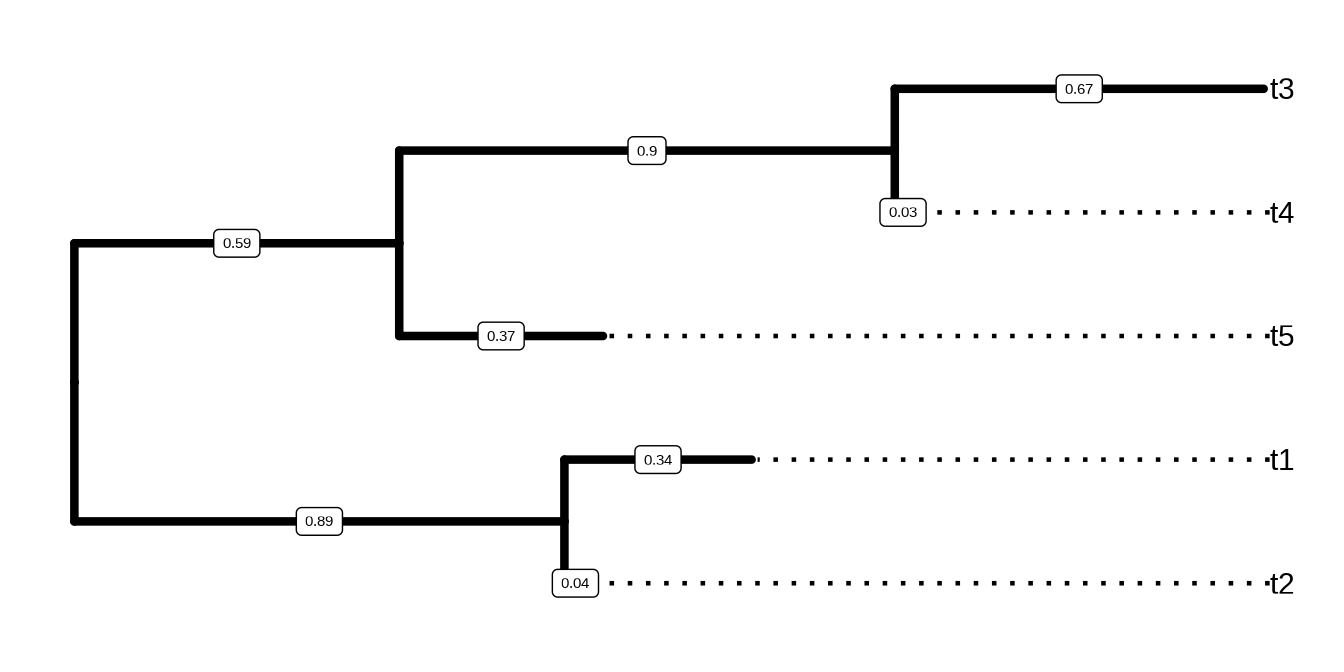
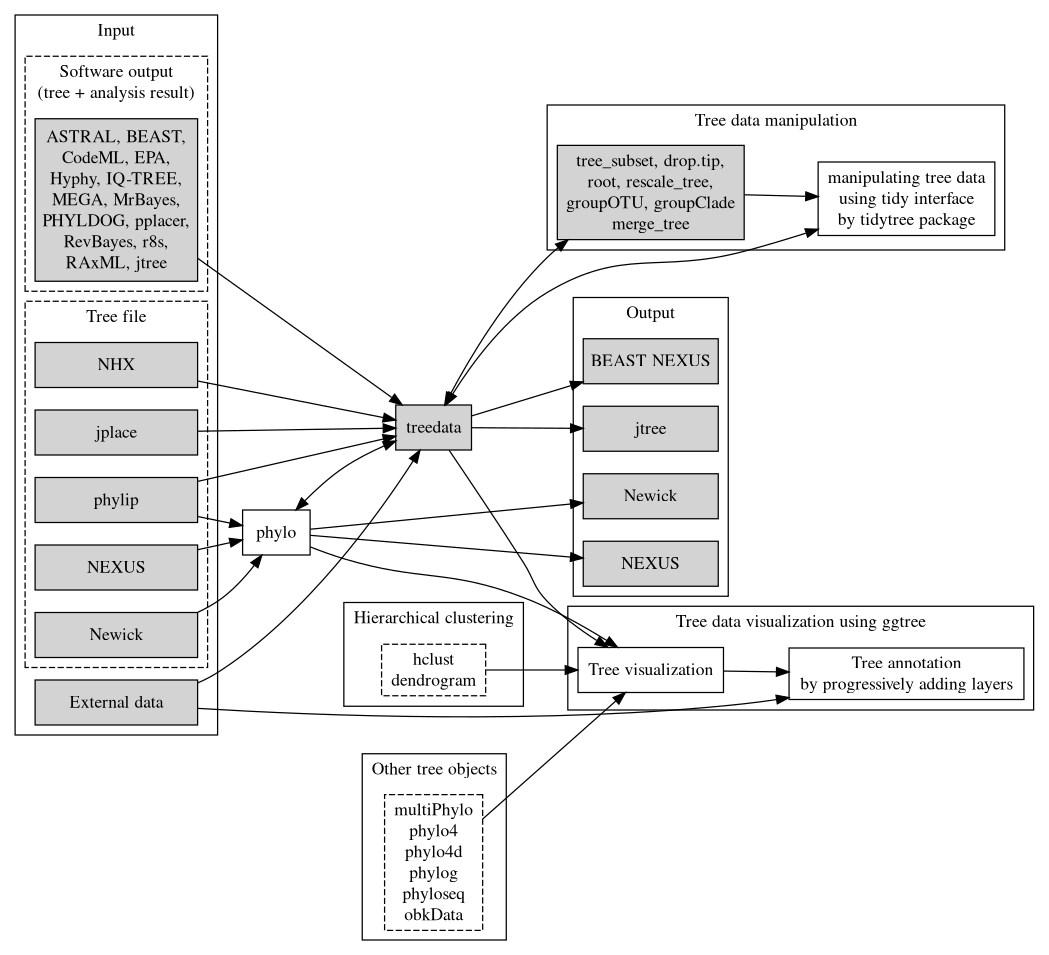
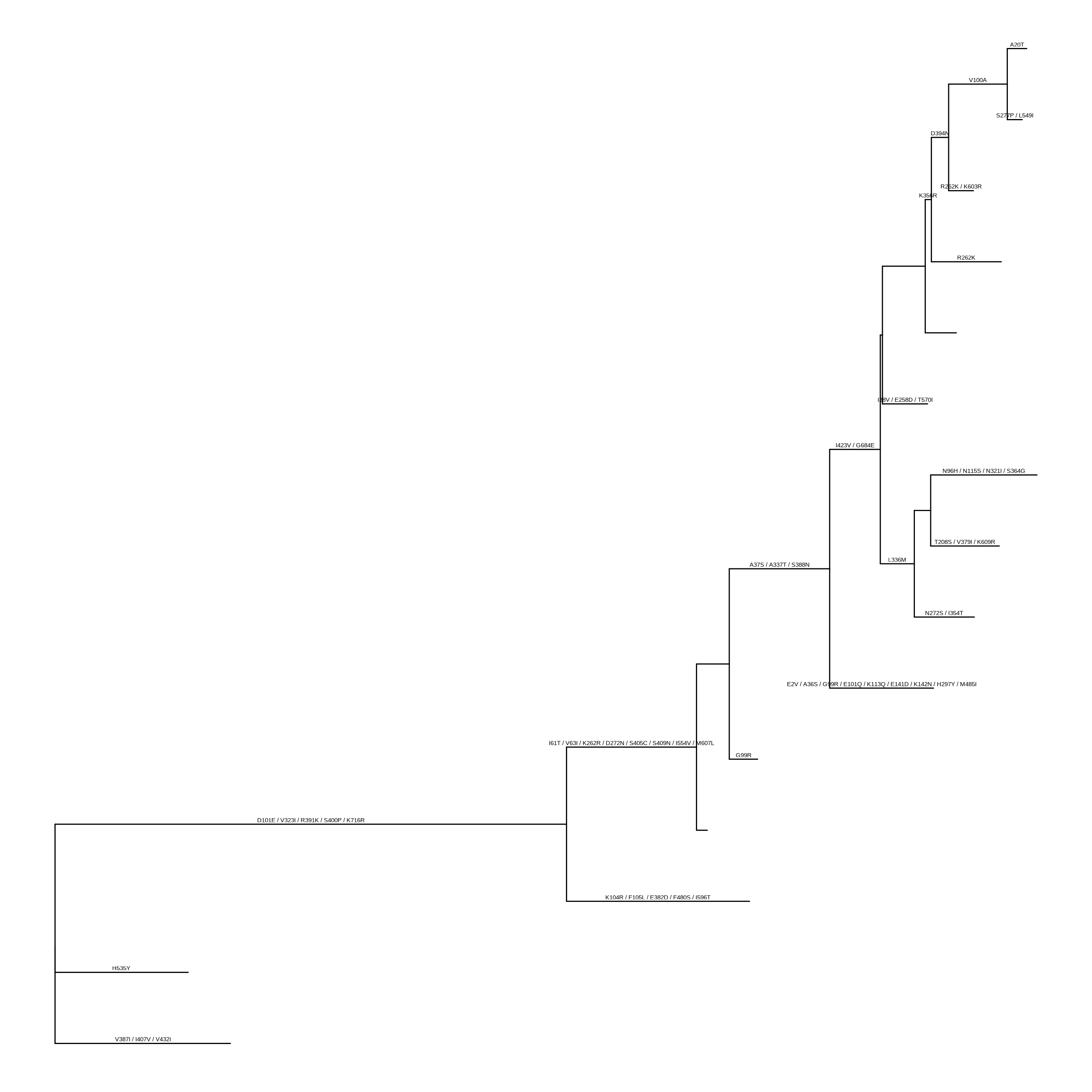
As phylogenetic trees are growing in their application to identify patterns in an evolutionary context, more different disciplines are employing phylogenetic trees in their research. For example, spatial ecologists may map the geographical positions of the organisms to their phylogenetic trees to understand the biogeography of the species (Schön et al., 2015); disease epidemiologists may incorporate the pathogen sampling time and locations into the phylogenetic analysis to infer the disease transmission dynamics in spatiotemporal space (He et al., 2013); microbiologists may determine the pathogenicity of different pathogen strains and map them into their phylogenetic trees to identify the genetic determinants of the pathogenicity (Bosi et al., 2016); genomic scientists may use the phylogenetic trees to help taxonomically classify their metagenomic sequence data (Gupta & Sharma, 2015). A robust tool such as treeio to import and map different types of data into the phylogenetic tree is important to facilitate these phylogenetics-related research, or ‘phylodynamics’. Such tools could also help integrate different meta-data (time, geography, genotype, epidemiological information) and analysis results (selective pressure, evolutionary rates) at the highest level and provide a comprehensive understanding of the study organisms. In the field of influenza research, there have been such attempts of studying the phylodynamics of the influenza virus by mapping different meta-data and analysis results on the same phylogenetic tree and evolutionary timescale (Lam et al., 2015).
Arenas, M. (2015). Trends in substitution models of molecular evolution.
Frontiers in Genetics,
6.
https://doi.org/10.3389/fgene.2015.00319
Berger, S. A., Krompass, D., & Stamatakis, A. (2011). Performance,
Accuracy, and
Web Server for
Evolutionary Placement of
Short Sequence Reads under
Maximum Likelihood.
Systematic Biology, 291–302.
https://doi.org/10.1093/sysbio/syr010
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., & Palsson, B. Ø. (2016). Comparative genome-scale modelling of
Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity.
Proceedings of the National Academy of Sciences of the United States of America,
113(26), E3801–E3809.
https://doi.org/10.1073/pnas.1523199113
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., Suchard, M. A., Rambaut, A., & Drummond, A. J. (2014).
BEAST 2:
A Software Platform for
Bayesian Evolutionary Analysis.
PLoS Comput Biol,
10(4), e1003537.
https://doi.org/10.1371/journal.pcbi.1003537
Boussau, B., Szöllősi, G. J., Duret, L., Gouy, M., Tannier, E., & Daubin, V. (2013). Genome-scale coestimation of species and gene trees.
Genome Research,
23(2), 323–330.
https://doi.org/10.1101/gr.141978.112
Felsenstein, J. (1978). Cases in which
Parsimony or
Compatibility Methods will be
Positively Misleading.
Systematic Biology,
27(4), 401–410.
https://doi.org/10.1093/sysbio/27.4.401
Felsenstein, J. (1981).
Evolutionary trees from DNA sequences: A maximum likelihood approach.
Journal of Molecular Evolution,
17(6), 368–376.
Felsenstein, J. (1989). PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics, 5, 164–166.
Fitch, W. M. (1971). Toward
Defining the
Course of
Evolution:
Minimum Change for a
Specific Tree Topology.
Systematic Zoology,
20(4), 406–416.
https://doi.org/10.2307/2412116
Goldman, N., & Yang, Z. (1994).
A codon-based model of nucleotide substitution for protein-coding DNA sequences.
Molecular Biology and Evolution,
11(5), 725–736.
Gupta, A., & Sharma, V. K. (2015). Using the taxon-specific genes for the taxonomic classification of bacterial genomes.
BMC Genomics,
16(1).
https://doi.org/10.1186/s12864-015-1542-0
He, Y.-Q., Chen, L., Xu, W.-B., Yang, H., Wang, H.-Z., Zong, W.-P., Xian, H.-X., Chen, H.-L., Yao, X.-J., Hu, Z.-L., Luo, M., Zhang, H.-L., Ma, H.-W., Cheng, J.-Q., Feng, Q.-J., & Zhao, D.-J. (2013). Emergence,
Circulation, and
Spatiotemporal Phylogenetic Analysis of
Coxsackievirus A6- and
Coxsackievirus A10-
Associated Hand,
Foot, and
Mouth Disease Infections from 2008 to 2012 in
Shenzhen,
China.
Journal of Clinical Microbiology,
51(11), 3560–3566.
https://doi.org/10.1128/JCM.01231-13
Höhna, S., Heath, T. A., Boussau, B., Landis, M. J., Ronquist, F., & Huelsenbeck, J. P. (2014). Probabilistic graphical model representation in phylogenetics.
Systematic Biology,
63(5), 753–771.
https://doi.org/10.1093/sysbio/syu039
Höhna, S., Landis, M. J., Heath, T. A., Boussau, B., Lartillot, N., Moore, B. R., Huelsenbeck, J. P., & Ronquist, F. (2016).
RevBayes:
Bayesian Phylogenetic Inference Using Graphical Models and an
Interactive Model-
Specification Language.
Systematic Biology,
65(4), 726–736.
https://doi.org/10.1093/sysbio/syw021
Huelsenbeck, J. P., & Ronquist, F. (2001).
MRBAYES: Bayesian inference of phylogenetic trees.
Bioinformatics (Oxford, England),
17(8), 754–755.
Kumar, S., Stecher, G., & Tamura, K. (2016).
MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets.
Molecular Biology and Evolution,
33(7), 1870–1874.
https://doi.org/10.1093/molbev/msw054
Lam, T. T.-Y., Zhou, B., Wang, J., Chai, Y., Shen, Y., Chen, X., Ma, C., Hong, W., Chen, Y., Zhang, Y., Duan, L., Chen, P., Jiang, J., Zhang, Y., Li, L., Poon, L. L. M., Webby, R. J., Smith, D. K., Leung, G. M., … Zhu, H. (2015). Dissemination, divergence and establishment of
H7N9 influenza viruses in
China.
Nature,
522(7554), 102–105.
https://doi.org/10.1038/nature14348
Lemmon, A. R., & Moriarty, E. C. (2004). The importance of proper model assumption in bayesian phylogenetics.
Systematic Biology,
53(2), 265–277.
https://doi.org/10.1080/10635150490423520
Maddison, D. R., Swofford, D. L., Maddison, W. P., & Cannatella, D. (1997). Nexus:
An Extensible File Format for
Systematic Information.
Systematic Biology,
46(4), 590–621.
https://doi.org/10.1093/sysbio/46.4.590
Matsen, F. A., Kodner, R. B., & Armbrust, E. V. (2010). Pplacer: Linear time maximum-likelihood and bayesian phylogenetic placement of sequences onto a fixed reference tree.
BMC Bioinformatics,
11(1), 538.
https://doi.org/10.1186/1471-2105-11-538
Paradis, E., Claude, J., & Strimmer, K. (2004).
APE:
Analyses of
Phylogenetics and
Evolution in
R language.
Bioinformatics,
20(2), 289–290.
https://doi.org/10.1093/bioinformatics/btg412
Pond, S. L. K., Frost, S. D. W., & Muse, S. V. (2005).
HyPhy: Hypothesis testing using phylogenies.
Bioinformatics (Oxford, England),
21(5), 676–679.
https://doi.org/10.1093/bioinformatics/bti079
R Core Team. (2016).
R: A language and environment for statistical computing. R Foundation for Statistical Computing.
https://www.R-project.org/
Retief, J. D. (2000).
Phylogenetic analysis using PHYLIP.
Methods in Molecular Biology (Clifton, N.J.),
132, 243–258.
Sanderson, M. J. (2003). r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock.
Bioinformatics,
19(2), 301–302.
https://doi.org/10.1093/bioinformatics/19.2.301
Schliep, K. P. (2011). Phangorn: Phylogenetic analysis in
R.
Bioinformatics,
27(4), 592–593.
https://doi.org/10.1093/bioinformatics/btq706
Schmidt, H. A., Strimmer, K., Vingron, M., & Haeseler, A. von. (2002).
TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing.
Bioinformatics (Oxford, England),
18(3), 502–504.
Schön, I., Shearn, R., Martens, K., Koenders, A., & Halse, S. (2015). Age and origin of
Australian Bennelongia (
Crustacea,
Ostracoda).
Hydrobiologia,
750(1), 125–146.
https://doi.org/10.1007/s10750-014-2159-z
Stamatakis, A. (2014).
RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies.
Bioinformatics,
30(9), 1312–1313.
https://doi.org/10.1093/bioinformatics/btu033
Wang, L.-G., Lam, T. T.-Y., Xu, S., Dai, Z., Zhou, L., Feng, T., Guo, P., Dunn, C. W., Jones, B. R., Bradley, T., Zhu, H., Guan, Y., Jiang, Y., & Yu, G. (2020). Treeio: An r package for phylogenetic tree input and output with richly annotated and associated data.
Molecular Biology and Evolution,
37(2), 599–603.
https://doi.org/10.1093/molbev/msz240
Wilgenbusch, J. C., & Swofford, D. (2003). Inferring evolutionary trees with
PAUP*.
Current Protocols in Bioinformatics,
Chapter 6, Unit 6.4.
https://doi.org/10.1002/0471250953.bi0604s00
Yang, Z. (2007).
PAML 4:
Phylogenetic Analysis by
Maximum Likelihood.
Molecular Biology and Evolution,
24(8), 1586–1591.
https://doi.org/10.1093/molbev/msm088
Yu, G. (2020). Using ggtree to visualize data on tree-like structures.
Current Protocols in Bioinformatics,
69(1), e96.
https://doi.org/10.1002/cpbi.96
Yu, G., Lam, T. T.-Y., Zhu, H., & Guan, Y. (2018). Two methods for mapping and visualizing associated data on phylogeny using ggtree.
Molecular Biology and Evolution,
35(12), 3041–3043.
https://doi.org/10.1093/molbev/msy194
Yu, G., Smith, D. K., Zhu, H., Guan, Y., & Lam, T. T.-Y. (2017). Ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data.
Methods in Ecology and Evolution,
8(1), 28–36.
https://doi.org/10.1111/2041-210X.12628
Zmasek, C. M., & Eddy, S. R. (2001).
ATV: Display and manipulation of annotated phylogenetic trees.
Bioinformatics,
17(4), 383–384.
https://doi.org/10.1093/bioinformatics/17.4.383