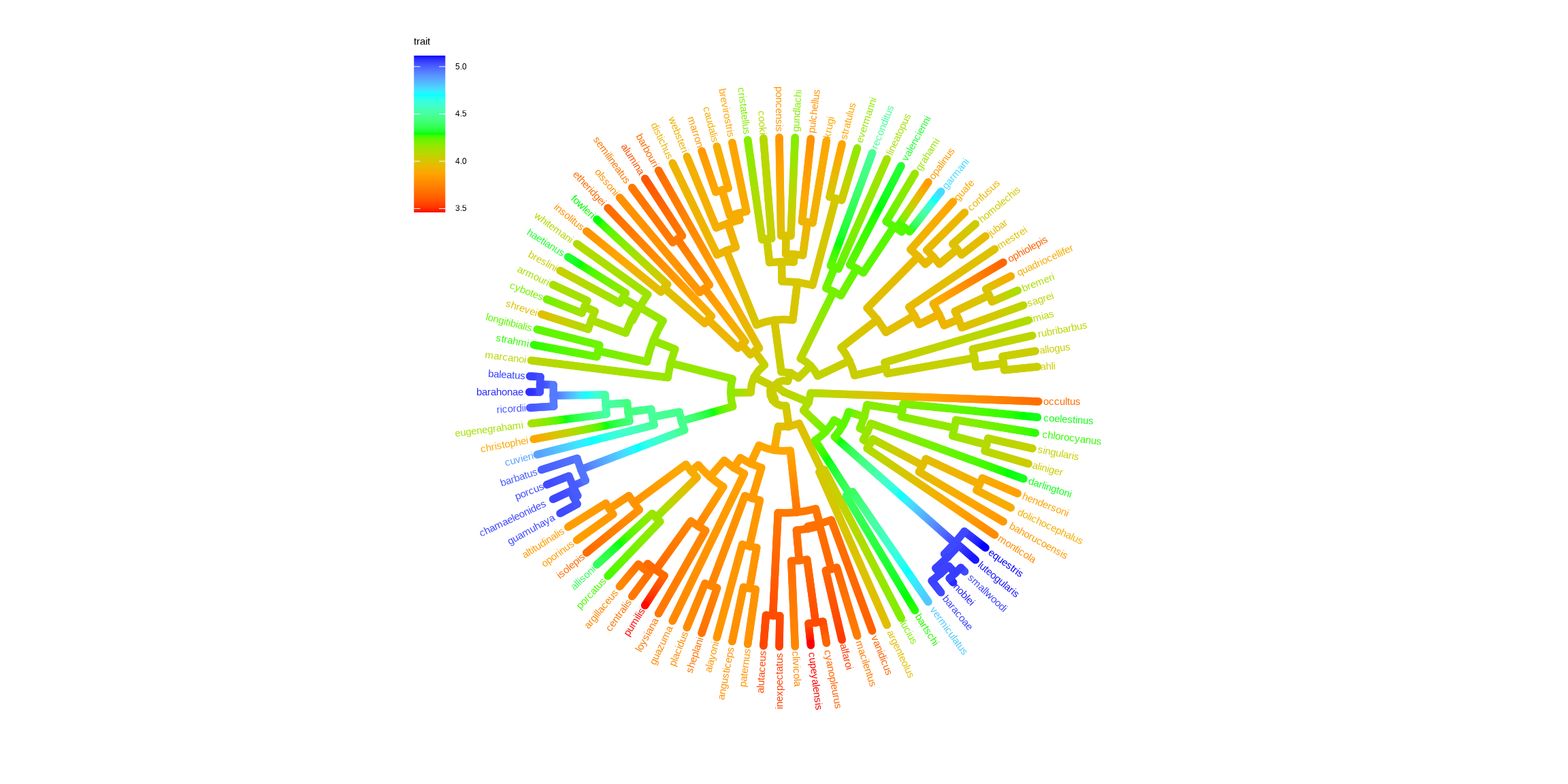
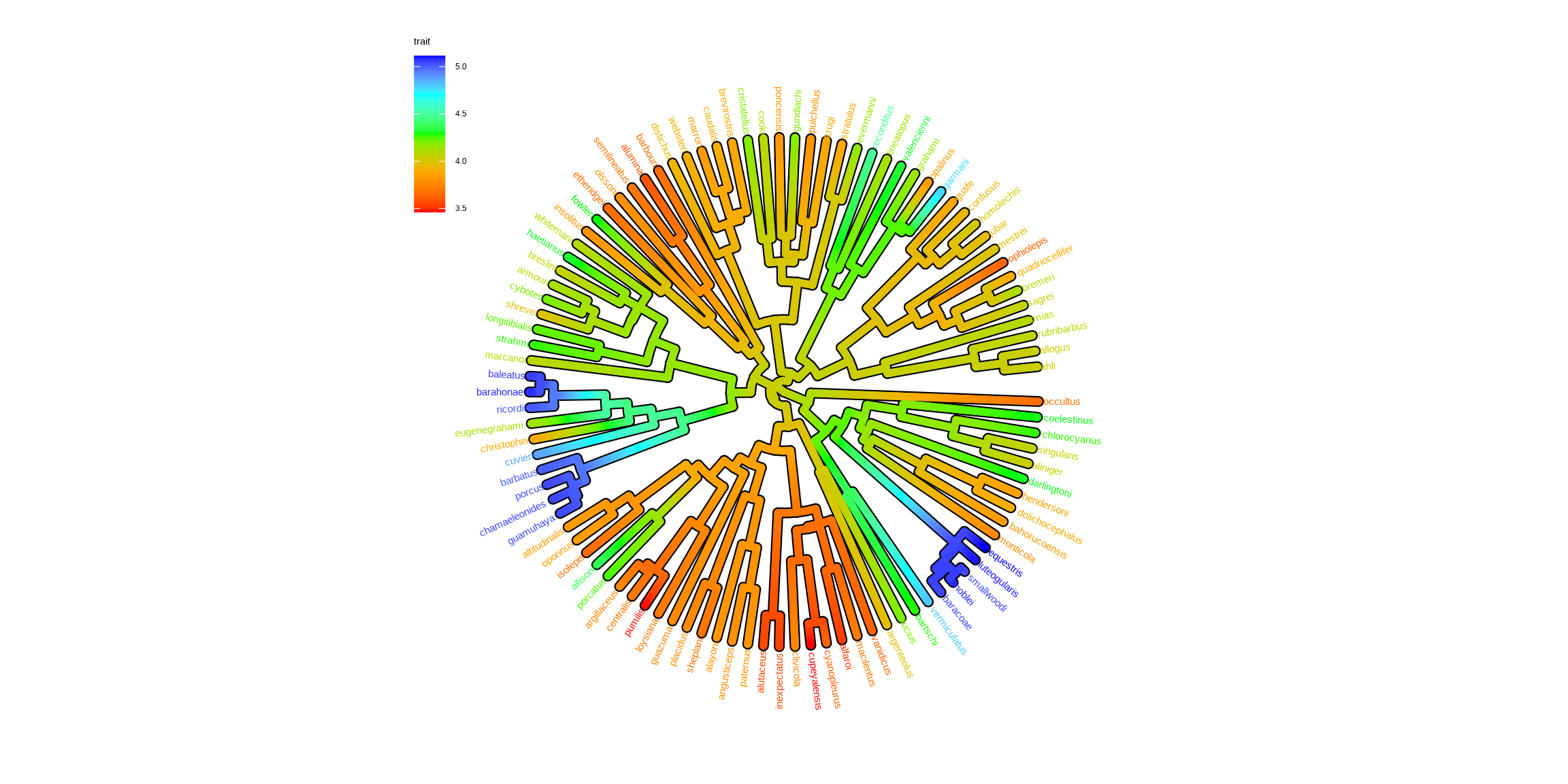
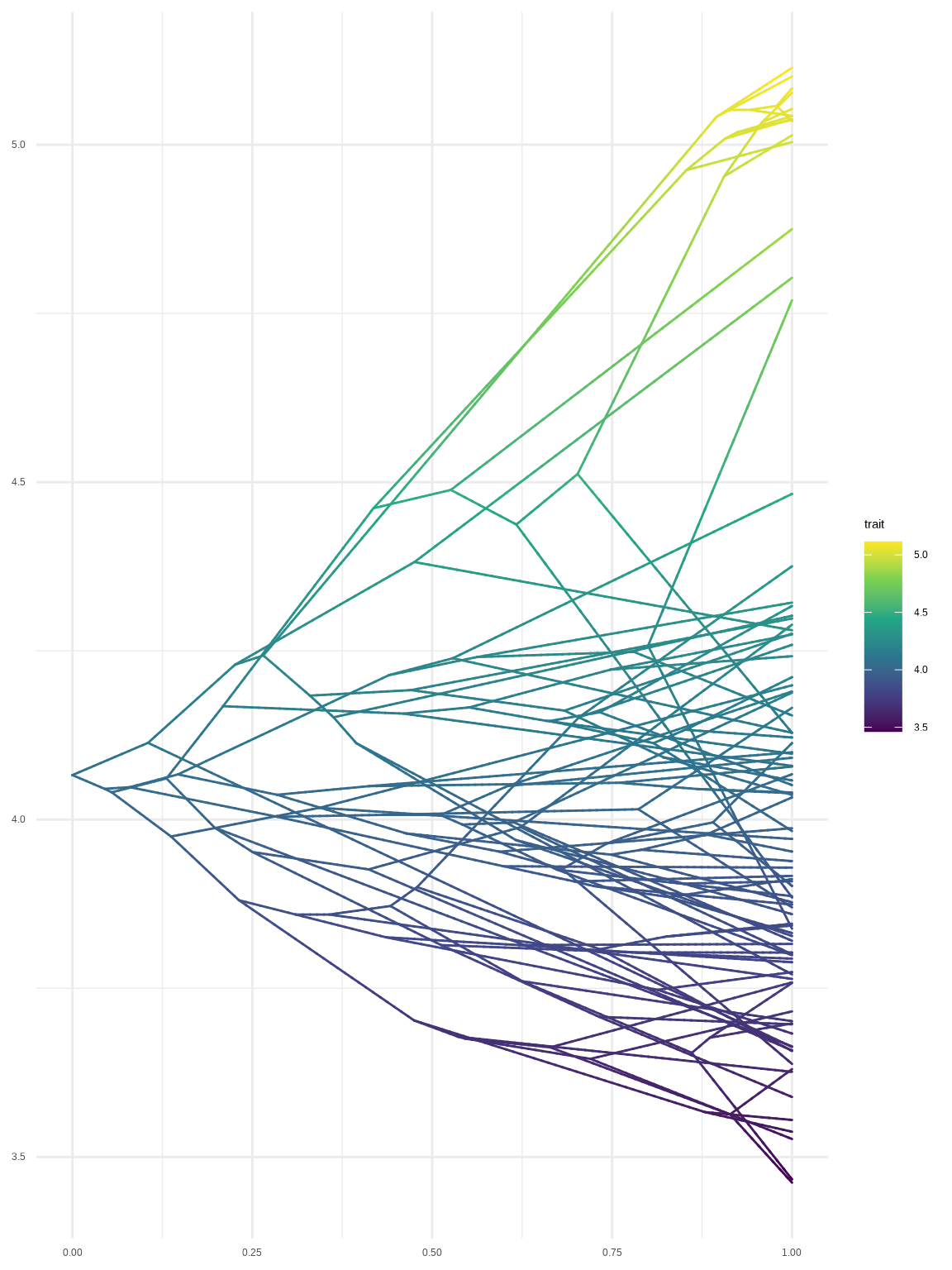
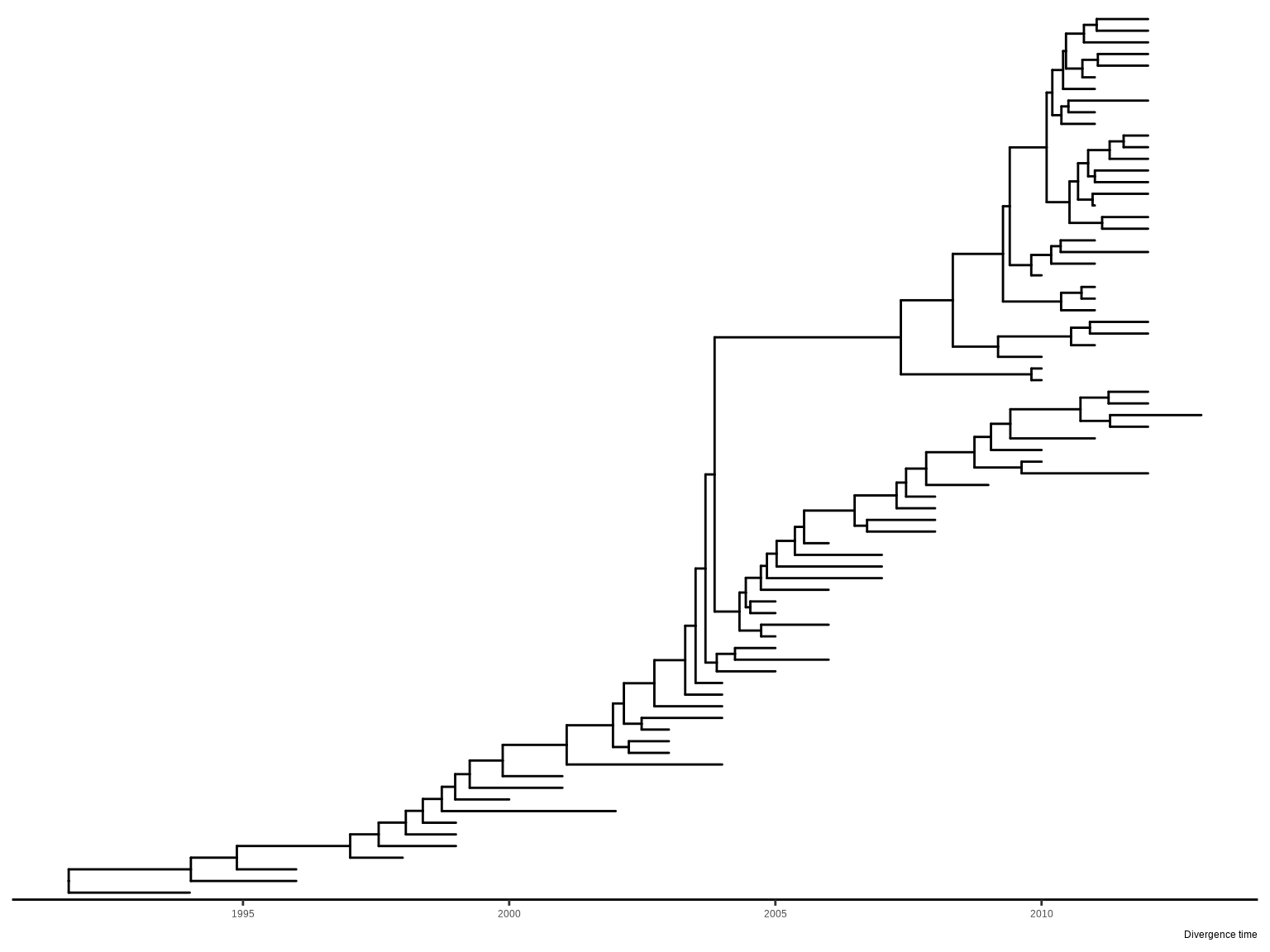
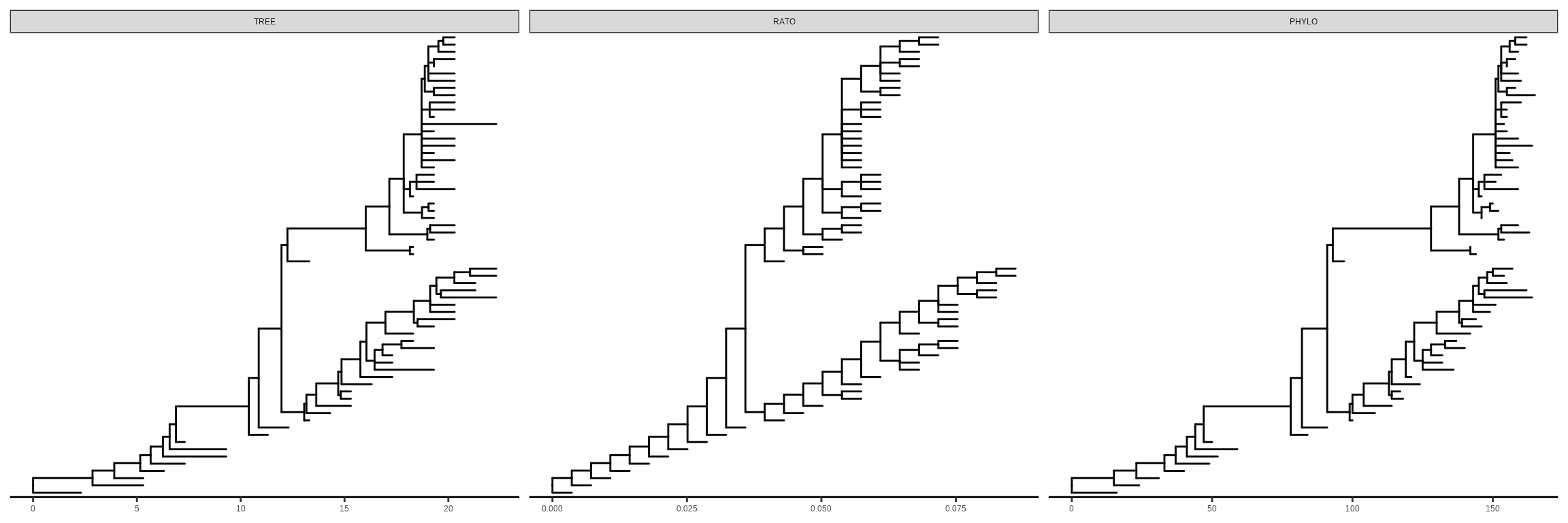
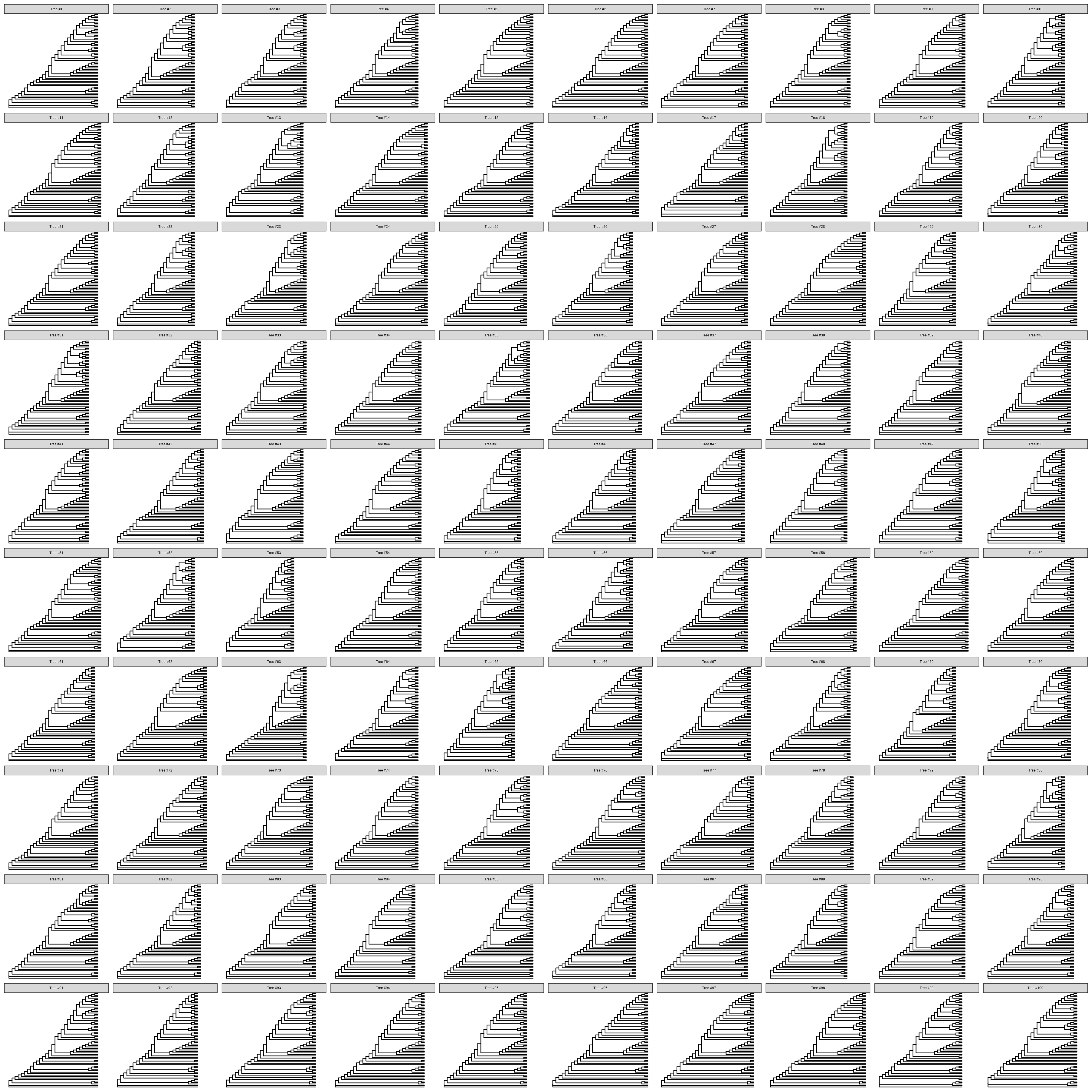
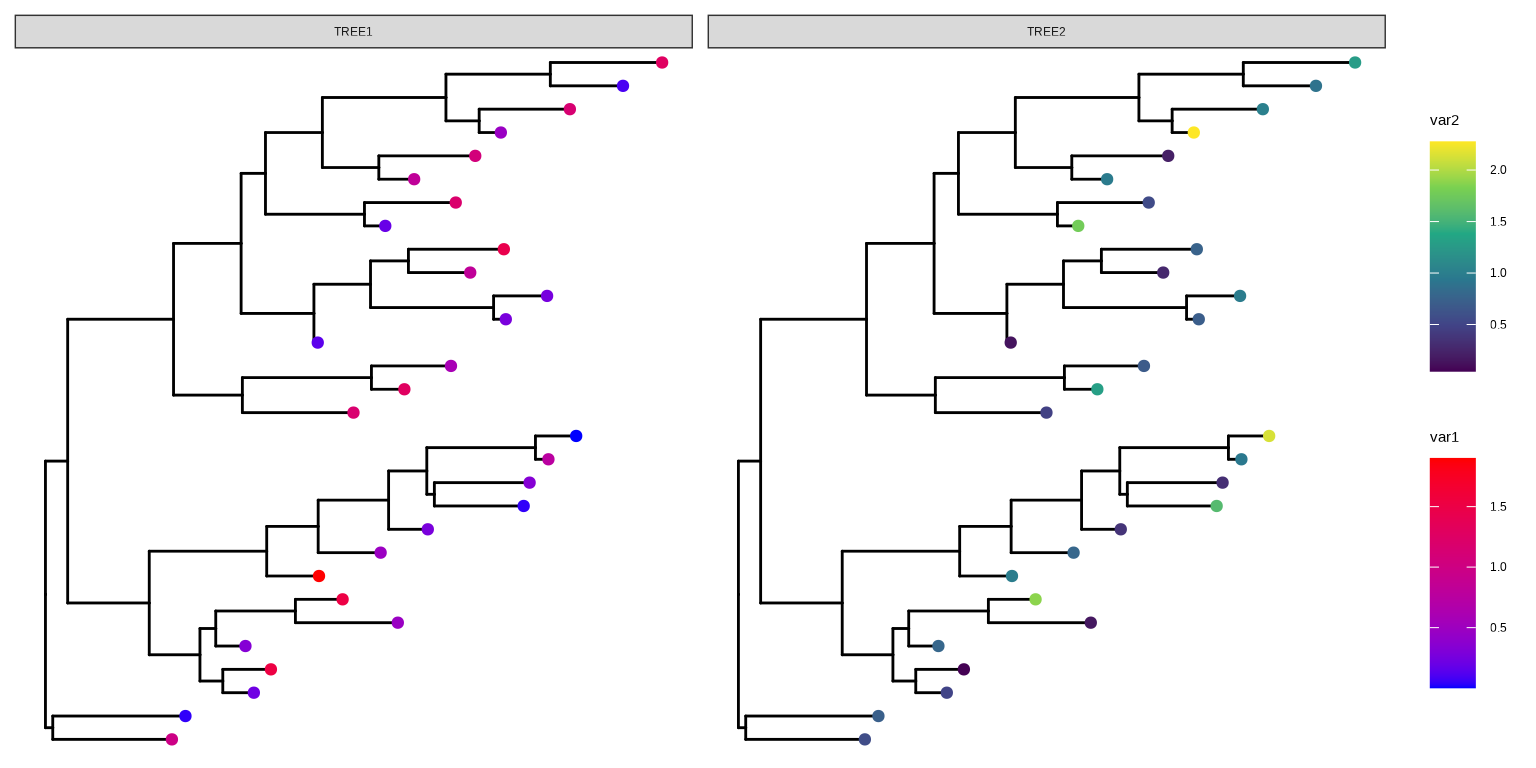
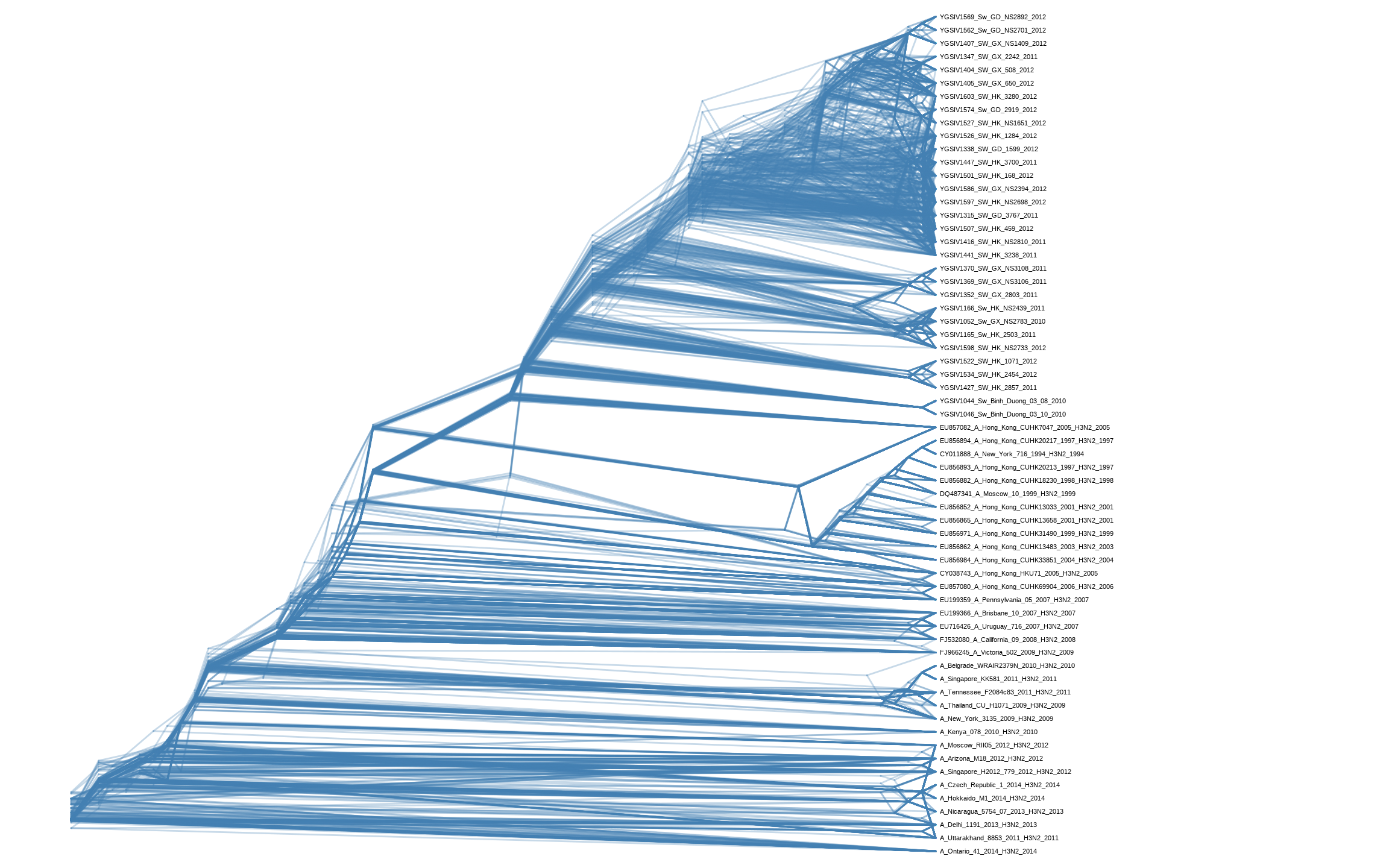
Chevenet, F., Brun, C., Bañuls, A.-L., Jacq, B., & Christen, R. (2006).
TreeDyn: Towards dynamic graphics and annotations for analyses of trees.
BMC Bioinformatics,
7, 439.
https://doi.org/10.1186/1471-2105-7-439
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., … Zhang, J. (2004). Bioconductor: Open software development for computational biology and bioinformatics.
Genome Biology,
5(10), R80.
https://doi.org/10.1186/gb-2004-5-10-r80
He, Z., Zhang, H., Gao, S., Lercher, M. J., Chen, W.-H., & Hu, S. (2016). Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees.
Nucleic Acids Research,
44(W1), W236–241.
https://doi.org/10.1093/nar/gkw370
Huson, D. H., & Scornavacca, C. (2012). Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks.
Systematic Biology,
61(6), 1061–1067.
https://doi.org/10.1093/sysbio/sys062
Jombart, T., Aanensen, D. M., Baguelin, M., Birrell, P., Cauchemez, S., Camacho, A., Colijn, C., Collins, C., Cori, A., Didelot, X., Fraser, C., Frost, S., Hens, N., Hugues, J., Höhle, M., Opatowski, L., Rambaut, A., Ratmann, O., Soubeyrand, S., … Ferguson, N. (2014).
OutbreakTools: A new platform for disease outbreak analysis using the
R software.
Epidemics,
7, 28–34.
https://doi.org/10.1016/j.epidem.2014.04.003
Lam, T. T.-Y., Hon, C.-C., Lemey, P., Pybus, O. G., Shi, M., Tun, H. M., Li, J., Jiang, J., Holmes, E. C., & Leung, F. C.-C. (2012). Phylodynamics of
H5N1 avian influenza virus in
Indonesia.
Molecular Ecology,
21(12), 3062–3077.
https://doi.org/10.1111/j.1365-294X.2012.05577.x
Letunic, I., & Bork, P. (2007). Interactive
Tree Of Life (
iTOL): An online tool for phylogenetic tree display and annotation.
Bioinformatics,
23(1), 127–128.
https://doi.org/10.1093/bioinformatics/btl529
Liang, H., Lam, T. T.-Y., Fan, X., Chen, X., Zeng, Y., Zhou, J., Duan, L., Tse, M., Chan, C.-H., Li, L., Leung, T.-Y., Yip, C.-H., Cheung, C.-L., Zhou, B., Smith, D. K., Poon, L. L.-M., Peiris, M., Guan, Y., & Zhu, H. (2014). Expansion of genotypic diversity and establishment of 2009
H1N1 pandemic-origin internal genes in pigs in
China.
Journal of Virology, JVI.01327–14.
https://doi.org/10.1128/JVI.01327-14
McMurdie, P. J., & Holmes, S. (2013). Phyloseq: An
R package for reproducible interactive analysis and graphics of microbiome census data.
PloS One,
8(4), e61217.
https://doi.org/10.1371/journal.pone.0061217
Neher, R. A., Bedford, T., Daniels, R. S., Russell, C. A., & Shraiman, B. I. (2016). Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses.
Proceedings of the National Academy of Sciences,
113(12), E1701–E1709.
https://doi.org/10.1073/pnas.1525578113
Page, R. D. M. (2002). Visualizing phylogenetic trees using
TreeView.
Current Protocols in Bioinformatics,
Chapter 6, Unit 6.2.
https://doi.org/10.1002/0471250953.bi0602s01
Paradis, E., Claude, J., & Strimmer, K. (2004).
APE:
Analyses of
Phylogenetics and
Evolution in
R language.
Bioinformatics,
20(2), 289–290.
https://doi.org/10.1093/bioinformatics/btg412
R Core Team. (2016).
R: A language and environment for statistical computing. R Foundation for Statistical Computing.
https://www.R-project.org/
Retief, J. D. (2000).
Phylogenetic analysis using PHYLIP.
Methods in Molecular Biology (Clifton, N.J.),
132, 243–258.
Wang, L.-G., Lam, T. T.-Y., Xu, S., Dai, Z., Zhou, L., Feng, T., Guo, P., Dunn, C. W., Jones, B. R., Bradley, T., Zhu, H., Guan, Y., Jiang, Y., & Yu, G. (2020). Treeio: An r package for phylogenetic tree input and output with richly annotated and associated data.
Molecular Biology and Evolution,
37(2), 599–603.
https://doi.org/10.1093/molbev/msz240
Wickham, H. (2016).
ggplot2: Elegant graphics for data analysis. Springer.
http://ggplot2.org
Wilgenbusch, J. C., & Swofford, D. (2003). Inferring evolutionary trees with
PAUP*.
Current Protocols in Bioinformatics,
Chapter 6, Unit 6.4.
https://doi.org/10.1002/0471250953.bi0604s00
Wilkinson, L., Wills, D., Rope, D., Norton, A., & Dubbs, R. (2005). The Grammar of Graphics (2nd edition). Springer.
Yu, G., Lam, T. T.-Y., Zhu, H., & Guan, Y. (2018). Two methods for mapping and visualizing associated data on phylogeny using ggtree.
Molecular Biology and Evolution,
35(12), 3041–3043.
https://doi.org/10.1093/molbev/msy194
Yu, G., Smith, D. K., Zhu, H., Guan, Y., & Lam, T. T.-Y. (2017). Ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data.
Methods in Ecology and Evolution,
8(1), 28–36.
https://doi.org/10.1111/2041-210X.12628