3 Coarse-graining of large single-cell data into metacells using SuperCell
Here, we use the pbmc4k dataset as presented in Batch correction.
3.1 RunSuperCell
The RunSuperCell() function is a wrapper function to run SuperCell.
## class: SingleCellExperiment
## dim: 33694 746
## metadata(3): hvgmethod hvgcols SuperCell
## assays(2): counts logcounts
## rownames(33694): RP11-34P13.3 FAM138A ... AC213203.1
## FAM231B
## rowData names(9): ENSEMBL_ID Symbol_TENx ... p.value
## FDR
## colnames: NULL
## colData names(1): size
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):The output of RunSuperCell() is a SingleCellExperiment object that stores gene expression matrix of metacells.
We can use it for downstram analysis.
post_process <- function(sce) {
sce <- NormalizeData(sce)
sce <- FindVariableFeatures(sce)
sce <- ScaleData(sce)
sce <- runPCA(sce, subset_row = VariableFeatures(sce), exprs_values = "scaled")
sce <- FindNeighbors(sce, dims = 1:10)
sce <- FindClusters(sce)
sce <- RunUMAP(sce)
return(sce)
}
sce_sc <- post_process(sce_sc)
library(ggsc)
sc_dim(sce_sc, reduction="UMAP") + sc_dim_geom_label()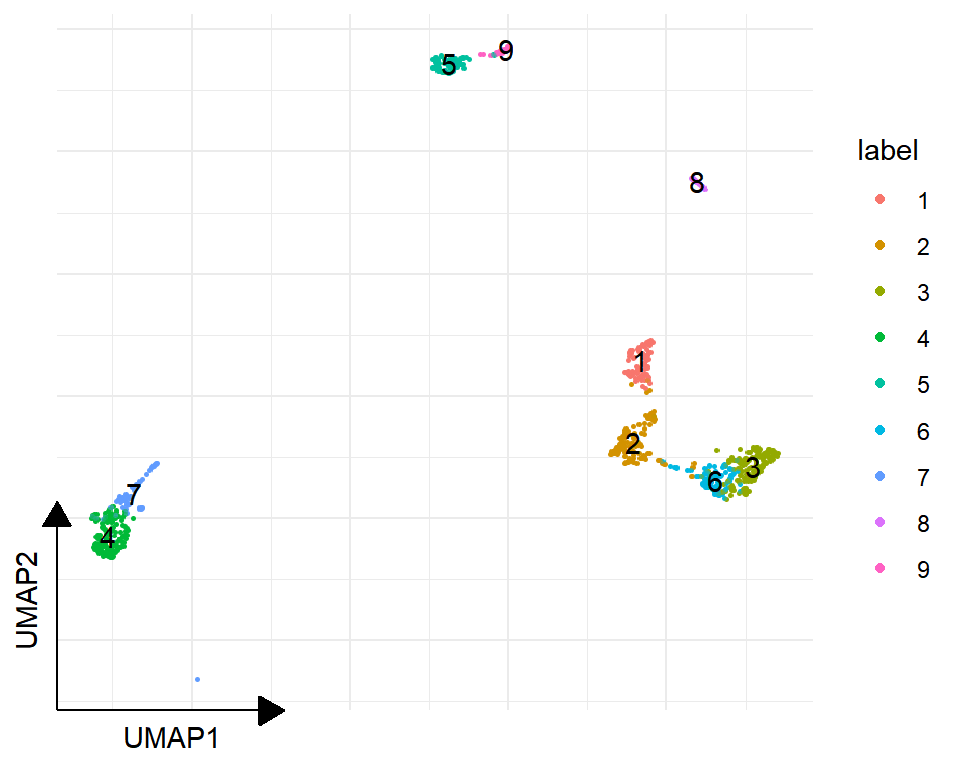
3.2 Estimate SuperCell purity
pbmc4k2 <- post_process(pbmc4k)
SC <- metadata(sce_sc)$SuperCell
purity <- SuperCell::supercell_purity(pbmc4k2$label, SC$membership, method = 'entropy')
head(purity)## 1 2 3 4 5 6
## 0 0 0 0 0 0## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.00000 0.00000 0.00000 0.03568 0.00000 0.69315