1 Importing Tree with Data
1.1 Overview of Phylogenetic Tree Construction
Phylogenetic trees are used to describe genealogical relationships among a group of organisms, which can be constructed based on the genetic sequences of the organisms. A rooted phylogenetic tree represents a model of evolutionary history depicted by ancestor-descendant relationships between tree nodes and clustering of ‘sister’ or ‘cousin’ organisms at a different level of relatedness, as illustrated in Figure 1.1. In infectious disease research, phylogenetic trees are usually built from the pathogens’ gene or genome sequences to show which pathogen sample is genetically closer to another sample, providing insights into the underlying unobserved epidemiologic linkage and the potential source of an outbreak.

FIGURE 1.1: Components of a phylogenetic tree. External nodes (green circles), also called ‘tips’, represent actual organisms sampled and sequenced (e.g., viruses in infectious disease research). They are the ‘taxa’ in the terminology of evolutionary biology. The internal nodes (blue circles) represent hypothetical ancestors for the tips. The root (red circle) is the common ancestor of all species in the tree. The horizontal lines are branches and represent evolutionary changes (gray number) measured in a unit of time or genetic divergence. The bar at the bottom provides the scale of these branch lengths.
A phylogenetic tree can be constructed from genetic sequences using distance-based methods or character-based methods. Distance-based methods, including the unweighted pair group method with arithmetic means (UPGMA) and the Neighbor-joining (NJ), are based on the matrix of pairwise genetic distances calculated between sequences. The character-based methods, including maximum parsimony (MP) (Fitch, 1971), maximum likelihood (ML) (Felsenstein, 1981), and Bayesian Markov Chain Monte Carlo (BMCMC) method (Rannala & Yang, 1996), are based on a mathematical model that describes the evolution of genetic characters and searches for the best phylogenetic tree according to their optimality criteria.
The MP method assumes that the evolutionary change is rare and minimizes the amount of character-state changes (e.g., number of DNA substitutions). The criterion is similar to Occam’s razor, that the simplest hypothesis that can explain the data is the best hypothesis. Unweighted parsimony assumes mutations across different characters (nucleotides or amino acids) are equally likely, while the weighted method assumes the unequal likelihood of mutations (e.g., the third codon position is more liable than other codon positions; and the transition mutations have a higher frequency than transversion). The concept of the MP method is straightforward and intuitive, which is a probable reason for its popularity amongst biologists who care more about the research question rather than the computational details of the analysis. However, this method has several disadvantages, in particular, the tree inference can be biased by the well-known systematic error called long-branch attraction (LBA) that incorrectly infer distantly related lineages as closely related (Felsenstein, 1978). This is because the MP method poorly takes into consideration of many sequence evolution factors (e.g., reversals and convergence) that are hardly observable from the existing genetic data.
The maximum likelihood (ML) method and Bayesian Markov Chain Monte Carlo (BMCMC) method are the two most commonly used methods in phylogenetic tree construction and are most often used in scientific publications. ML and BMCMC methods require a substitution model of sequence evolution. Different sequence data have different substitution models to formulate the evolutionary process of DNA, codon and amino acid. There are several models for nucleotide substitution, including JC69, K2P, F81, HKY, and GTR (Arenas, 2015). These models can be used in conjunction with the rate variation across sites (denoted as +\(\Gamma\))) (Yang, 1994) and the proportion of invariable sites (denoted as +I) (Shoemaker & Fitch, 1989). Previous research (Lemmon & Moriarty, 2004) had suggested that misspecification of substitution model might bias phylogenetic inference. Procedural testing for the best-fit substitution model is recommended.
The optimal criterion of the ML method is to find the tree that maximizes the likelihood given the sequence data. The procedure of the ML method is simple: calculating the likelihood of a tree and optimizing its topology and branches (and the substitution model parameters, if not fixed) until the best tree is found. Heuristic search, such as those implemented in PhyML and RAxML, is often used to find the best tree based on the likelihood criterion. The Bayesian method finds the tree that maximizes posterior probability by sampling trees through MCMC based on the given substitution model. One of the advantages of BMCMC is that parameter variance and tree topological uncertainty, included by the posterior clade probability, can be naturally and conveniently obtained from the sampling trees in the MCMC process. Moreover, the influence of topological uncertainty on other parameter estimates is also naturally integrated into the BMCMC phylogenetic framework.
In a simple phylogenetic tree, data associated with the tree branches/nodes could be the branch lengths (indicating genetic or time divergence) and lineage supports such as bootstrap values estimated from bootstrapping procedure or posterior clade probability summarized from the sampled trees in the BMCMC analysis.
1.2 Phylogenetic Tree Formats
There are several file formats designed to store phylogenetic trees and the data associated with the nodes and branches. The three commonly used formats are Newick3, NEXUS (Maddison et al., 1997), and Phylip (Felsenstein, 1989). Some formats (e.g., NHX) are extended from the Newick format. Newick and NEXUS formats are supported as input by most of the software in evolutionary biology, while some of the software tools output newer standard files (e.g., BEAST and MrBayes) by introducing new rules/data blocks for storing evolutionary inferences. In the other cases (e.g., PAML and r8s), output log files are only recognized by their own single software.
1.2.1 Newick tree format
The Newick tree format is the standard for representing trees in computer-readable form.

FIGURE 1.2: A sample tree for demonstrating Newick text to encode tree structure. Tips were aligned to the right-hand side and branch lengths were labeled on the middle of each branch.
The rooted tree shown in Figure 1.2 can be represented by the following sequence of characters as a Newick tree text.
((t2:0.04,t1:0.34):0.89,(t5:0.37,(t4:0.03,t3:0.67):0.9):0.59); The tree text ends with a semicolon. Internal nodes are represented by a pair of matched parentheses. Between the parentheses are descendant nodes of that node. For instance (t2:0.04,t1:0.34) represents the parent node of t2 and t1 that are the immediate descendants. Sibling nodes are separated by a comma and tips are represented by their names. A branch length (from the parent node to child node) is represented by a real number after the child node and is preceded by a colon. Singular data (e.g., bootstrap values) associated with internal nodes or branches may be encoded as node labels and represented by the simple text/numbers before the colon.
Newick tree format was developed by Meacham in 1984 for the Phylogeny Inference Package or PHYLIP (Retief, 2000) package. Newick format is now the most widely used tree format and used by PHYLIP, PAUP* (Wilgenbusch & Swofford, 2003), TREE-PUZZLE (Schmidt et al., 2002), MrBayes, and many other applications. Phylip tree format contains Phylip multiple sequence alignment (MSA) with a corresponding Newick tree text that was built based on the MSA sequences in the same file.
1.2.2 NEXUS tree format
The NEXUS format (Maddison et al., 1997) incorporates Newick tree text with related information organized into separated units known as blocks. A NEXUS block has the following structure:
#NEXUS
...
BEGIN characters;
...
END;For example, the above example tree can be saved as the following NEXUS format:
#NEXUS
[R-package APE, Wed Jul 13 17:10:18 2022]
BEGIN TAXA;
DIMENSIONS NTAX = 5;
TAXLABELS
t2
t1
t5
t4
t3
;
END;
BEGIN TREES;
TRANSLATE
1 t2,
2 t1,
3 t5,
4 t4,
5 t3
;
TREE * UNTITLED = [&R]
((1:0.04,2:0.34):0.89,(3:0.37,(4:0.03,5:0.67):0.9):0.59);
END;Comments can be placed using square brackets. Some blocks can be recognized by most of the programs including TAXA (contains information of taxa), DATA (contains data matrix, e.g., sequence alignment), and TREE (contains a phylogenetic tree, i.e., Newick tree text). Notably, blocks can be very diverse and some of them are only recognized by one particular program. For example NEXUS file exported by PAUP* has a paup block that contains PAUP* commands, whereas FigTree exports the NEXUS file with a figtree block that contains visualization settings. NEXUS organizes different types of data into different blocks, whereas programs that support reading NEXUS can parse some blocks they recognized and ignore those they could not. This is a good mechanism to allow different programs to use the same format without crashing when unsupported types of data are present. Notably, most of the programs only support parsing TAXA, DATA, and TREE blocks; therefore, a program/platform that could generically read all kinds of data blocks from the NEXUS would be useful for phylogenetic data integration.
The DATA block is widely used to store sequence alignment. For this purpose, the user can store tree and sequence data in Phylip format which are essentially Phylip multiple sequence alignment and Newick tree text, respectively. It is used in PHYLIP.
1.2.3 New Hampshire eXtended format
Newick, NEXUS, and phylip are mainly designed to store phylogenetic trees and basic singular data associated with internal nodes or branches. In addition to the singular data annotation at branches and nodes (mentioned above), New Hampshire eXtended (NHX) format, which is based on Newick (also called New Hampshire), was developed to introduce tags to associate multiple data fields with the tree nodes (both internal nodes and tips). Tags are placed after branch length and must be wrapped between [&&NHX and ] which makes it possible to be compatible with NEXUS format as it defined characters between [ and ] as comments. NHX is also the output format of PHYLDOG (Boussau et al., 2013) and RevBayes (Höhna et al., 2016). A Tree Viewer (ATV) (Zmasek & Eddy, 2001) is a java tool that supports displaying annotation data stored in NHX format, but this package is no longer maintained.
Here is a sample tree from NHX definition document4:
(((ADH2:0.1[&&NHX:S=human], ADH1:0.11[&&NHX:S=human]):0.05
[&&NHX:S=primates:D=Y:B=100],ADHY:0.1[&&NHX:S=nematode],
ADHX:0.12[&&NHX:S=insect]):0.1[&&NHX:S=metazoa:D=N],(ADH4:0.09
[&&NHX:S=yeast],ADH3:0.13[&&NHX:S=yeast],ADH2:0.12[&&NHX:S=yeast],
ADH1:0.11[&&NHX:S=yeast]):0.1[&&NHX:S=Fungi])[&&NHX:D=N];1.2.4 Jplace format
To store the Next Generation Sequencing (NGS) short reads mapped onto a phylogenetic tree (for metagenomic classification), Matsen (Matsen et al., 2012) proposed jplace format for such phylogenetic placements. Jplace format is based on JSON and contains four keys: tree, fields, placements, metadata, and version. The tree value contains tree text extended from Newick tree format by putting the edge label in brackets (if available) after branch length and putting the edge number in curly braces after the edge label. The fields value contains header information of placement data. The value of placements is a list of pqueries. Each pquery contains two keys: p for placements and n for name or nm for names with multiplicity. The value of p is a list of placement for pqueries.
Here is a jplace sample file:
{
"tree": "(((((((A:4{1},B:4{2}):6{3},C:5{4}):8{5},D:6{6}):
3{7},E:21{8}):10{9},((F:4{10},G:12{11}):14{12},H:8{13}):
13{14}):13{15},((I:5{16},J:2{17}):30{18},(K:11{19},
L:11{20}):2{21}):17{22}):4{23},M:56{24});",
"placements": [
{"p":[24, -61371.300778, 0.333344, 0.000003, 0.003887],
"n":["AA"]
},
{"p":[[1, -61312.210786, 0.333335, 0.000001, 0.000003],
[2, -61322.210823, 0.333322, 0.000003, 0.000003],
[3, -61352.210823, 0.333322, 0.000961, 0.000003]],
"n":["BB"]
},
{"p":[[8, -61312.229128, 0.200011, 0.000001, 0.000003],
[9, -61322.229179, 0.200000, 0.000003, 0.000003],
[10, -61342.229223, 0.199992, 0.000003, 0.000003]],
"n":["CC"]
}
],
"metadata": {"info": "a jplace sample file"},
"version" : 2,
"fields": ["edge_num", "likelihood", "like_weight_ratio",
"distal_length", "pendant_length"
]
}Jplace is the output format of PPLACER (Matsen et al., 2010) and Evolutionary Placement Algorithm (EPA) (Berger et al., 2011). But these two programs do not contain tools to visualize placement results. PPLACER provides placeviz to convert jplace file to phyloXML or Newick formats which can be visualized by Archaeopteryx.
1.2.5 Software outputs
RAxML (Stamatakis, 2014) can output Newick format by storing the bootstrap values as internal node labels. Another way that RAxML supports is to place bootstrap value inside square brackets and after branch length. This could not be supported by most of the software that supports Newick format where square brackets will be ignored.
BEAST (Bouckaert et al., 2014) output is based on NEXUS, and it also introduces square brackets in the tree block to store evolutionary evidence inferred by BEAST. Inside brackets, curly braces may also be incorporated if feature values have a length of more than 1 (e.g., Highest Probability Density (HPD) or range of substitution rate). These brackets are placed between node and branch length (i.e., after label if exists and before the colon). The bracket is not defined in Newick format and is a reserved character for NEXUS comment. So this information will be ignored for standard NEXUS parsers.
Here is a sample TREE block of the BEAST output:
TREE * TREE1 = [&R] (((11[&length=9.47]:9.39,14[&length=6.47]:6.39)
[&length=25.72]:25.44,4[&length=9.14]:8.82)[&length=3.01]:3.1,
(12[&length=0.62]:0.57,(10[&length=1.6]:1.56,(7[&length=5.21]:5.19,
((((2[&length=3.3]:3.26,(1[&length=1.34]:1.32,(6[&length=0.85]:0.83,
13[&length=0.85]:0.83)[&length=2.5]:2.49)[&length=0.97]:0.94)
[&length=0.5]:0.5,9[&length=1.76]:1.76)[&length=2.41]:2.36,
8[&length=2.19]:2.11)[&length=0.27]:0.24,(3[&length=3.33]:3.31,
(15[&length=5.29]:5.27,5[&length=3.29]:3.27)[&length=1.04]:1.04)
[&length=1.98]:2.04)[&length=2.83]:2.84)[&length=5.39]:5.37)
[&length=2.02]:2)[&length=4.35]:4.36)[&length=0];BEAST output can contain many different evolutionary inferences, depending on the analysis models defined in BEAUTi for running. For example in molecular clock analysis, it contains rate, length, height, posterior and corresponding HPD and range for uncertainty estimation. Rate is the estimated evolutionary rate of the branch. Length is the length of the branch in years. Height is the time from node to root, while posterior is the Bayesian clade credibility value. The above example is the output tree of a molecular clock analysis and should contain these inferences. To save space, only the length estimation was shown above. Besides, Molecular Evolutionary Genetics Analysis (MEGA) (Kumar et al., 2016) also supports exporting trees in BEAST compatible Nexus format (see session 1.3.2).
MrBayes (Huelsenbeck & Ronquist, 2001) is a program that uses the Markov Chain Monte Carlo method to sample from the posterior probability distributions. Its output file annotates nodes and branches separately by two sets of square brackets. For example below, posterior clade probabilities for the node and branch length estimates for the branch:
tree con_all_compat = [&U] (8[&prob=1.0]:2.94e-1[&length_mean=2.9e-1],
10[&prob=1.0]:2.25e-1[&length_mean=2.2e-1],((((1[&prob=1.0]:1.43e-1
[&length_mean=1.4e-1],2[&prob=1.0]:1.92e-1[&length_mean=1.9e-1])[&prob=1.0]:
1.24e-1[&length_mean=1.2e-1],9[&prob=1.0]:2.27e-1[&length_mean=2.2e-1])
[&prob=1.0]:1.72e-1[&length_mean=1.7e-1],12[&prob=1.0]:5.11e-1
[&length_mean=5.1e-1])[&prob=1.0]:1.76e-1[&length_mean=1.7e-1],
(((3[&prob=1.0]:5.46e-2[&length_mean=5.4e-2],(6[&prob=1.0]:1.03e-2
[&length_mean=1.0e-2],7[&prob=1.0]:7.13e-3[&length_mean=7.2e-3])[&prob=1.0]:
6.93e-2[&length_mean=6.9e-2])[&prob=1.0]:6.03e-2[&length_mean=6.0e-2],
(4[&prob=1.0]:6.27e-2[&length_mean=6.2e-2],5[&prob=1.0]:6.31e-2
[&length_mean=6.3e-2])[&prob=1.0]:6.07e-2[&length_mean=6.0e-2])[&prob=1.0]:,
1.80e-1[&length_mean=1.8e-1]11[&prob=1.0]:2.37e-1[&length_mean=2.3e-1])
[&prob=1.0]:4.05e-1[&length_mean=4.0e-1])[&prob=1.0]:1.16e+000
[&length_mean=1.162699558201079e+000])[&prob=1.0][&length_mean=0];To save space, most of the inferences were removed and only contains prob for clade probability and length_mean for mean value of branch length. The full version of this file also contains prob_stddev, prob_range, prob(percent), prob+-sd for probability inferences and length_median, length_95%_HPD for every branch.
The BEAST and MrBayes outputs are expected to be parsed without inferences (dropped as comments) by software that supports NEXUS. FigTree supports parsing BEAST, and MrBayes outputs with inferences that can be used to display or annotate on the tree. But from there, extracting these data for further analysis is still challenging.
HyPhy (Pond et al., 2005) could do a number of phylogenetic analyses, including ancestral sequence reconstruction. For ancestral sequence reconstruction, these sequences and the Newick tree text are stored in NEXUS format as the major analysis output. It did not completely follow the NEXUS definition and only put the ancestral node labels in TAXA instead of the external node label. The MATRIX block contains sequence alignment of ancestral nodes which cannot be referred back to the tree stored in the TREES block since it does not contain node labels. Here is the sample output (to save space, only the first 72bp of alignment are shown):
#NEXUS
[
Generated by HYPHY 2.0020110620beta(MP) for MacOS(Universal Binary)
on Tue Dec 23 13:52:34 2014
]
BEGIN TAXA;
DIMENSIONS NTAX = 13;
TAXLABELS
'Node1' 'Node2' 'Node3' 'Node4' 'Node5' 'Node12' 'Node13' 'Node15'
'Node18' 'Node20' 'Node22' 'Node24' 'Node26' ;
END;
BEGIN CHARACTERS;
DIMENSIONS NCHAR = 2148;
FORMAT
DATATYPE = DNA
GAP=-
MISSING=?
NOLABELS
;
MATRIX
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATTGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAATGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
ATGGAAGACTTTGTGCGACAGTGCTTCAATCCAATGATCGTCGAGCTTGCGGAAAAGGCAATGAAAGAATAT
END;
BEGIN TREES;
TREE tree = (K,N,(D,(L,(J,(G,((C,(E,O)),(H,(I,(B,(A,(F,M)))))))))));
END;There are other applications that output rich information text that also contains phylogenetic trees with associated data. For example r8s (Sanderson, 2003) output three trees in its log file, namely TREE, RATE, and PHYLO for branches scaled by time, substitution rate, and absolute substitutions, respectively.
Phylogenetic Analysis by Maximum Likelihood (PAML) (Yang, 2007) is a package of programs for phylogenetic analyses of DNA or protein sequences. Two main programs, BASEML and CODEML, implement a variety of models. BASEML estimates tree topology, branch lengths, and substitution parameters using a number of nucleotide substitution models available, including JC69, K80, F81, F84, HKY85, T92, TN93, and GTR. CODEML estimates synonymous and non-synonymous substitution rates, likelihood ratio test of positive selection under codon substitution models (Goldman & Yang, 1994).
BASEML outputs mlb file that contains input sequence (taxa) alignment and a phylogenetic tree with branch length as well as substitution model and other parameters estimated. The supplementary result file, rst, contains sequence alignment (with ancestral sequence if it performs reconstruction of ancestral sequences) and Naive Empirical Bayes (NBE) probabilities that each site in the alignment evolved. CODEML outputs mlc file that contains tree structure and estimation of synonymous and non-synonymous substitution rates. CODEML also outputs a supplementary result file, rst, that is similar to BASEML except that site is defined as a codon instead of a nucleotide. Parsing these files can be tedious and would oftentimes need a number of post-processing steps and require expertise in programming (e.g., with Python5 or Perl6).
Introducing square brackets is quite common for storing extra information, including RAxML to store bootstrap value, NHX format for annotation, jplace for edge label, and BEAST for evolutionary estimation, etc. But the positions to place square brackets are not consistent in different software and the contents employ different rules for storing associated data, these properties make it difficult to parse associated data. For most of the software, they will just ignore square brackets and only parse the tree structure if the file is compatible. Some of them contain invalid characters (e.g., curly braces in tree field of jplace format), and even the tree structure can’t be parsed by standard parsers.
It is difficult to extract useful phylogeny/taxon-related information from the different analysis outputs produced by various evolutionary inference software, for displaying on the same phylogenetic tree and for further analysis. FigTree supports BEAST output, but not for most of the other software outputs that contain evolutionary inferences or associated data. For those output-rich text files (e.g., r8s, PAML, etc.), the tree structure cannot be parsed by any tree viewing software and users need expertise to manually extract the phylogenetic tree and other useful result data from the output file. However, such manual operation is slow and error-prone.
It was not easy to retrieve phylogenetic trees with evolutionary data from different analysis outputs of commonly used software in the field. Some of them (e.g., PAML output and jplace file) without software or programming library to support parsing the file, while others (e.g., BEAST and MrBayes output) can be parsed without evolutionary inferences as they are stored in square brackets that will be omitted as a comment by most of the software. Although FigTree support visualizing evolutionary statistics inferred by BEAST and MrBayes, extracting these data for further analysis is not supported. Different software packages implement different algorithms for different analyses (e.g., PAML for dN/dS, HyPhy for ancestral sequences, and BEAST for skyline analysis). Therefore, in encountering the genomic sequence data, there is a desired need to efficiently and flexibly integrate different analysis inference results for comprehensive understanding, comparison, and further analysis. This motivated us to develop the programming library to parse the phylogenetic trees and data from various sources.
1.3 Getting Tree Data with treeio
Phylogenetic trees are commonly used to present evolutionary relationships of species. Information associated with taxon species/strains may be further analyzed in the context of the evolutionary history depicted by the phylogenetic tree. For example, the host information of the influenza virus strains in the tree could be studied to understand the host range of a virus lineage. Moreover, such meta-data (e.g., isolation host, time, location, etc.) directly associated with taxon strains are also often subjected to further evolutionary or comparative phylogenetic models and analyses, to infer their dynamics associated with the evolutionary or transmission processes of the virus. All these meta-data or other phenotypic or experimental data are stored either as the annotation data associated with the nodes or branches and are often produced in inconsistent format by different analysis programs.
Getting trees into R is still limited. Newick and Nexus can be imported by several packages, including ape, phylobase. NeXML format can be parsed by RNeXML. However, analysis results from widely used software packages in this field are not well supported. SIMMAP output can be parsed by phyext2 and phytools. Although PHYLOCH can import BEAST and MrBayes output, only internal node attributes were parsed and tip attributes were ignored7. Many other software outputs are mainly required programming expertise to import the tree with associated data. Linking external data, including experimental and clinical data, to phylogeny is another obstacle for evolution biologists.
To fill the gap that most of the tree formats or software outputs cannot be parsed within the same software/platform, an R package treeio (Wang et al., 2020) was developed for parsing various tree file formats and outputs from common evolutionary analysis software. The treeio package is developed with the R programming language (R Core Team, 2016). Not only the tree structure can be parsed, but also the associated data and evolutionary inferences, including NHX annotation, clock rate inferences (from BEAST or r8s (Sanderson, 2003) programs), synonymous and non-synonymous substitutions (from CODEML), and ancestral sequence construction (from HyPhy, BASEML or CODEML), etc.. Currently, treeio is able to read the following file formats: Newick, Nexus, New Hampshire eXtended format (NHX), jplace and Phylip as well as the data outputs from the following analysis programs: ASTRAL, BEAST, EPA, HyPhy, MEGA, MrBayes, PAML, PHYLDOG, PPLACER, r8s, RAxML and RevBayes, etc. This is made possible with the several parser functions developed in treeio (Table 1.1) (Wang et al., 2020).
| Parser function | Description |
|---|---|
| read.astral | parsing output of ASTRAL |
| read.beast | parsing output of BEAST |
| read.codeml | parsing output of CodeML (rst and mlc files) |
| read.codeml_mlc | parsing mlc file (output of CodeML) |
| read.fasta | parsing FASTA format sequence file |
| read.hyphy | parsing output of HYPHY |
| read.hyphy.seq | parsing ancestral sequences from HYPHY output |
| read.iqtree | parsing IQ-Tree Newick string, with the ability to parse SH-aLRT and UFBoot support values |
| read.jplace | parsing jplace file including the output of EPA and pplacer |
| read.jtree | parsing jtree format |
| read.mega | parsing MEGA Nexus output |
| read.mega_tabular | parsing MEGA tabular output |
| read.mrbayes | parsing output of MrBayes |
| read.newick | parsing Newick string, with the ability to parse node label as support values |
| read.nexus | parsing standard NEXUS file (re-exported from ape) |
| read.nhx | parsing NHX file including the output of PHYLDOG and RevBayes |
| read.paml_rst | parsing rst file (output of BaseML or CodeML) |
| read.phylip | parsing phylip file (phylip alignment + Newick string) |
| read.phylip.seq | parsing multiple sequence alignment from phylip file |
| read.phylip.tree | parsing newick string from phylip file |
| read.phyloxml | parsing phyloXML file |
| read.r8s | parsing output of r8s |
| read.raxml | parsing output of RAxML |
| read.tree | parsing newick string (re-exported from ape) |
The treeio package defines base
classes and functions for phylogenetic tree input and output. It is an
infrastructure that enables evolutionary evidence inferred by commonly
used software packages to be used in R. For instance, dN/dS values or
ancestral sequences inferred
by CODEML (Yang, 2007),
clade support values (posterior) inferred
by BEAST (Bouckaert et al., 2014) and short read placement
by EPA (Berger et al., 2011)
and PPLACER (Matsen et al., 2010). These pieces of
evolutionary evidence can be further analyzed in R and used to annotate
a phylogenetic tree using ggtree
(Yu et al., 2017). The growth of analysis tools and models introduces
a challenge to integrate different varieties of data and analysis results from
different sources for an integral analysis on the same phylogenetic tree
background. The treeio package (Wang et al., 2020)
provides a merge_tree function to allow combining tree data obtained from
different sources. In addition, treeio also enables external data to be linked to a phylogenetic tree structure.
After parsing, storage of the tree structure with associated data is made
through an S4 class, treedata, defined in the tidytree package. These parsed data
are mapped to the tree branches and nodes inside treedata object, so that they
can be efficiently used to visually annotate the tree
using ggtree (Yu et al., 2017) and ggtreeExtra (Xu, Dai, et al., 2021).
A programmable platform for phylogenetic data parsing, integration, and annotations as such makes us more easily to identify the evolutionary dynamics and correlation patterns (Figure 1.3) (Wang et al., 2020).
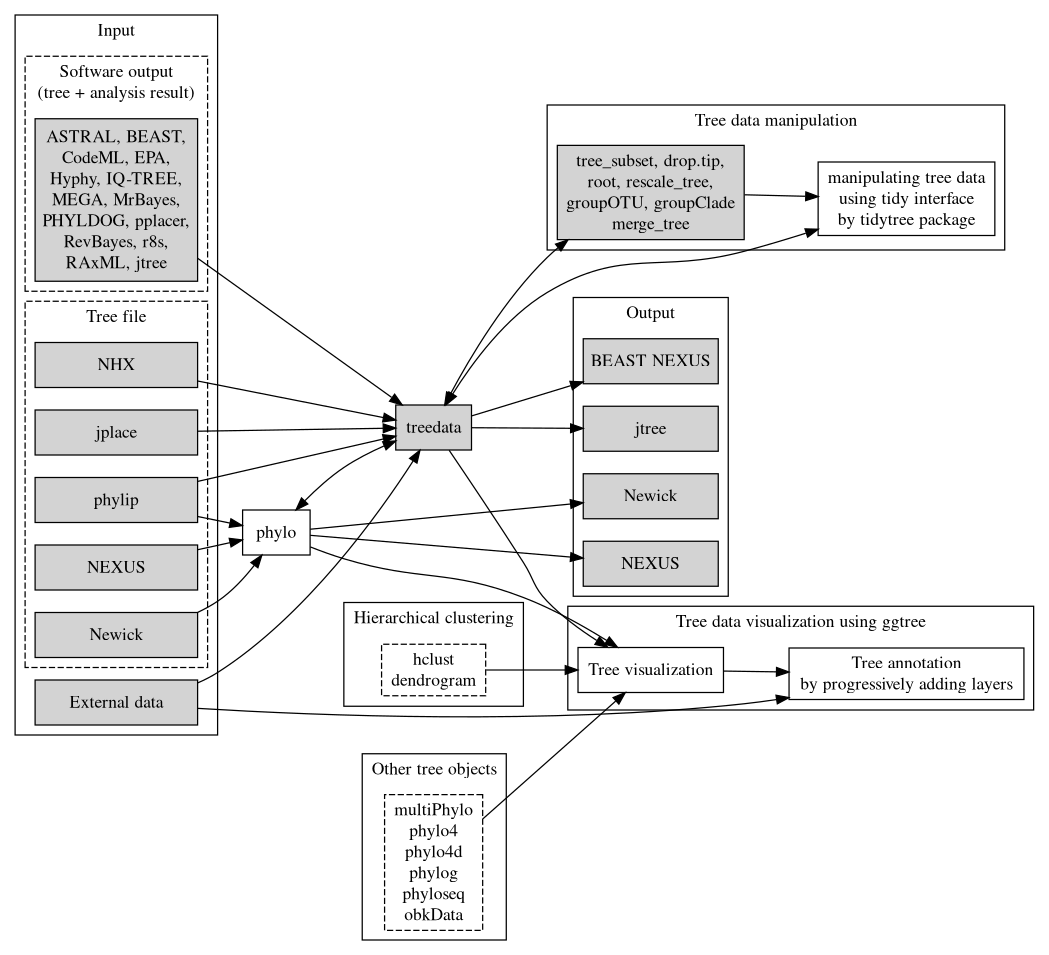
FIGURE 1.3: Overview of the treeio package and its relations with tidytree and ggtree. Treeio supports parsing a tree with data from a number of file formats and software outputs. A treedata object stores a phylogenetic tree with node/branch-associated data. Treeio provides several functions to manipulate a tree with data. Users can convert the treedata object into a tidy data frame (each row represents a node in the tree and each column represents a variable) and process the tree with data using the tidy interface implemented in tidytree. The tree can be extracted from the treedata object and exported to a Newick and NEXUS file or can be exported with associated data into a single file (either in the BEAST NEXUS or jtree format). Associated data stored in the treedata object can be used to annotate the tree using ggtree. In addition, ggtree supports a number of tree objects, including phyloseq for microbiome data and obkData for outbreak data. The phylo, multiPhylo (ape package), phylo4, phylo4d (phylobase package), phylog (ade4 package), phyloseq (phyloseq package), and obkData (OutbreakTools package) are tree objects defined by the R community to store tree with or without domain-specific data. All these tree objects as well as hierarchical clustering results (e.g., hclust and dendrogram objects) are supported by ggtree.
1.3.1 Overview of treeio
The treeio package (Wang et al., 2020) defined S4 classes for storing phylogenetic trees with diverse types of associated data or covariates from different sources including analysis outputs from different software packages. It also defined corresponding parser functions for parsing phylogenetic trees with annotation data and stored them as data objects in R for further manipulation or analysis (see Table 1.1). Several accessor functions were defined to facilitate accessing the tree annotation data, including get.fields for obtaining annotation features available in the tree object, get.placements for obtaining the phylogenetic placement results (i.e., the output of PPLACER, EPA, etc.), get.subs for obtaining the genetic substitutions from parent node to child node, and get.tipseq for getting the tip sequences.
The S3 class, phylo, which was defined in ape (Paradis et al., 2004) package, is widely used in R community and many packages. As treeio uses S4 class, to enable those available R packages to analyze the tree imported by treeio, treeio provides as.phylo function to convert treeio-generated tree object to phylo object that only contains tree structure without annotation data. In the other way, treeio also provides as.treedata function to convert phylo object with evolutionary analysis result (e.g., bootstrap values calculated by ape or ancestral states inferred by phangorn (Schliep, 2011) etc.) to be stored as a treedata S4 object, making it easy to map the data to the tree structure and to be visualized using ggtree (Yu et al., 2017).
To allow integration of different kinds of data in a phylogenetic tree, treeio (Wang et al., 2020) provides merge_tree function (details in section 2.2.1) for combining evolutionary statistics/evidence imported from different sources including those common tree files and outputs from analysis programs (Table 1.1). There is other information, such as sampling location, taxonomy information, experimental result, and evolutionary traits, etc. that are stored in separate files with the user-defined format. In treeio, we could read in these data from the users’ files using standard R IO functions, and attach them to the tree object by the full_join methods defined in tidytree and treeio packages (see also the %<+% operator defined in ggtree). After attaching, the data will become the attributes associated with nodes or branches, which can be compared with other data incorporated or can be visually displayed on the tree (Yu et al., 2018).
To facilitate storing complex data associated with the phylogenetic tree, treeio implemented write.beast and write.jtree functions to export a treedata object into a single file (see Chapter 3).
1.3.2 Function demonstration
1.3.2.1 Parsing BEAST output
file <- system.file("extdata/BEAST", "beast_mcc.tree", package="treeio")
beast <- read.beast(file)
beast## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/BEAST/beast_mcc.tree'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 14 internal nodes.
##
## Tip labels:
## A_1995, B_1996, C_1995, D_1987, E_1996, F_1997, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_median',
## 'height_range', 'length', 'length_0.95_HPD',
## 'length_median', 'length_range', 'posterior', 'rate',
## 'rate_0.95_HPD', 'rate_median', 'rate_range'.Since % is not a valid character in names, all the feature names that contain x% will convert to 0.x. For example, length_95%_HPD will be changed to length_0.95_HPD.
Not only tree structure but also all the features inferred by BEAST will be stored in the S4 object. These features can be used for tree annotation (Figure 5.8).
1.3.2.2 Parsing MEGA output
Molecular Evolutionary Genetics Analysis (MEGA) software (Kumar et al., 2016) supports exporting trees in three distinct formats: Newick, tabular, and Nexus. The Newick file can be parsed using the read.tree or read.newick functions. MEGA Nexus file is similar to BEAST Nexus and treeio (Wang et al., 2020) provides read.mega function to parse the tree.
file <- system.file("extdata/MEGA7", "mtCDNA_timetree.nex",
package = "treeio")
read.mega(file)## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/MEGA7/mtCDNA_timetree.nex'.
##
## ...@ phylo:
##
## Phylogenetic tree with 7 tips and 6 internal nodes.
##
## Tip labels:
## homo_sapiens, chimpanzee, bonobo, gorilla,
## orangutan, sumatran, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'branch_length', 'data_coverage', 'rate',
## 'reltime', 'reltime_0.95_CI', 'reltime_stderr'.The tabular output contains tree and associated information (divergence time in this example) in a tabular flat text file. The read.mega_tabular function can parse the tree with data simultaneously.
file <- system.file("extdata/MEGA7", "mtCDNA_timetree_tabular.txt",
package = "treeio")
read.mega_tabular(file) ## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/MEGA7/mtCDNA_timetree_tabular.txt'.
##
## ...@ phylo:
##
## Phylogenetic tree with 7 tips and 6 internal nodes.
##
## Tip labels:
## chimpanzee, bonobo, homo sapiens, gorilla,
## orangutan, sumatran, ...
## Node labels:
## , , demoLabel2, , ,
##
## Rooted; no branch lengths.
##
## with the following features available:
## 'RelTime', 'CI_Lower', 'CI_Upper', 'Rate', 'Data
## Coverage'.1.3.2.3 Parsing MrBayes output
Although the Nexus file generated by MrBayes is different from the output of BEAST, they are similar. The treeio package provides the read.mrbayes() which internally calls read.beast() to parse MrBayes outputs.
file <- system.file("extdata/MrBayes", "Gq_nxs.tre", package="treeio")
read.mrbayes(file)## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/MrBayes/Gq_nxs.tre'.
##
## ...@ phylo:
##
## Phylogenetic tree with 12 tips and 10 internal nodes.
##
## Tip labels:
## B_h, B_s, G_d, G_k, G_q, G_s, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'length_0.95HPD', 'length_mean', 'length_median',
## 'prob', 'prob_range', 'prob_stddev', 'prob_percent',
## 'prob+-sd'.1.3.2.4 Parsing PAML output
Phylogenetic Analysis by Maximum Likelihood (PAML) is a package of tools for phylogenetic analyses of DNA and protein sequences using maximum likelihood. Tree search algorithms are implemented in BASEML and CODEML. The read.paml_rst() function provided in treeio can parse rst file from BASEML and CODEML. The only difference is the space in the sequences.
For BASEML, every ten bases are separated by one space, while for CODEML, every three bases (triplet) are separated by one space.
brstfile <- system.file("extdata/PAML_Baseml", "rst", package="treeio")
brst <- read.paml_rst(brstfile)
brst## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/PAML_Baseml/rst'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## A, B, C, D, E, F, ...
## Node labels:
## 16, 17, 18, 19, 20, 21, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'subs', 'AA_subs'.Similarly, we can parse the rst file from CODEML.
crstfile <- system.file("extdata/PAML_Codeml", "rst", package="treeio")
## type can be one of "Marginal" or "Joint"
crst <- read.paml_rst(crstfile, type = "Joint")
crst## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/PAML_Codeml/rst'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## A, B, C, D, E, F, ...
## Node labels:
## 16, 17, 18, 19, 20, 21, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'subs', 'AA_subs'.Ancestral sequences inferred by BASEML or CODEML via marginal or joint ML reconstruction methods will be stored in the S4 object and mapped to tree nodes. treeio (Wang et al., 2020) will automatically determine the substitutions between the sequences at both ends of each branch. The amino acid substitution will also be determined by translating nucleotide sequences to amino acid sequences. These computed substitutions will also be stored in the S4 object for efficient tree annotation later (Figure 5.10).
CODEML infers selection
pressure and estimated dN/dS, dN and dS. These pieces of information are
stored in output file mlc, which can be parsed by the read.codeml_mlc() function.
mlcfile <- system.file("extdata/PAML_Codeml", "mlc", package="treeio")
mlc <- read.codeml_mlc(mlcfile)
mlc## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/PAML_Codeml/mlc'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## A, B, C, D, E, F, ...
## Node labels:
## 16, 17, 18, 19, 20, 21, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 't', 'N', 'S', 'dN_vs_dS', 'dN', 'dS', 'N_x_dN',
## 'S_x_dS'.The rst and mlc files can be parsed separately as demonstrated previously, they
can also be parsed together using the read.codeml() function.
## tree can be one of "rst" or "mlc" to specify
## using tree from which file as base tree in the object
ml <- read.codeml(crstfile, mlcfile, tree = "mlc")
ml## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/PAML_Codeml/rst',
## '/home/ygc/R/library/treeio/extdata/PAML_Codeml/mlc'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## A, B, C, D, E, F, ...
## Node labels:
## 16, 17, 18, 19, 20, 21, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'subs', 'AA_subs', 't', 'N', 'S', 'dN_vs_dS', 'dN',
## 'dS', 'N_x_dN', 'S_x_dS'.All the features in both rst and mlc files are imported into a single S4 object and hence are available for further annotation and visualization. For example, we can annotate and display both dN/dS (from mlc file) and amino acid substitutions (derived from rst file) on the same phylogenetic tree (Yu et al., 2017).
1.3.2.5 Parsing HyPhy output
Hypothesis testing using Phylogenies (HyPhy) is a software package for analyzing genetic sequences. Ancestral sequences inferred by HyPhy are
stored in the Nexus output file, which contains the tree topology and ancestral
sequences. To parse this data file, users can use the read.hyphy.seq() function.
ancseq <- system.file("extdata/HYPHY", "ancseq.nex", package="treeio")
read.hyphy.seq(ancseq)## 13 DNA sequences in binary format stored in a list.
##
## All sequences of same length: 2148
##
## Labels:
## Node1
## Node2
## Node3
## Node4
## Node5
## Node12
## ...
##
## Base composition:
## a c g t
## 0.335 0.208 0.237 0.220
## (Total: 27.92 kb)To map the sequences on the tree, users should also provide an internal-node-labeled tree. If users want to determine substitution, they need to also provide tip sequences. In this case, substitutions will be determined automatically, just as we parse the output of CODEML.
nwk <- system.file("extdata/HYPHY", "labelledtree.tree", package="treeio")
tipfas <- system.file("extdata", "pa.fas", package="treeio")
hy <- read.hyphy(nwk, ancseq, tipfas)
hy## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/HYPHY/labelledtree.tree'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## K, N, D, L, J, G, ...
## Node labels:
## Node1, Node2, Node3, Node4, Node5, Node12, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'subs', 'AA_subs'.1.3.2.6 Parsing r8s output
The r8s package uses parametric, semi-parametric, and non-parametric methods to relax the molecular clock to allow better estimations of divergence times and evolution rates (Sanderson, 2003). It outputs three trees in a log file, namely, TREE, RATO, and PHYLO for time tree, rate tree, and absolute substitution tree, respectively.
The time tree is scaled by divergence time, the rate tree is scaled by substitution rate and the absolute substitution tree is scaled by the absolute number of substitutions. After parsing the file, all these three trees are stored in a multiPhylo object (Figure 4.15).
r8s <- read.r8s(system.file("extdata/r8s", "H3_r8s_output.log", package="treeio"))
r8s## 3 phylogenetic trees1.3.2.7 Parsing output of RAxML bootstrap analysis
RAxML bootstrapping analysis
outputs a Newick tree text that is not standard, as it stores bootstrap values
inside square brackets after branch lengths. This file usually cannot be parsed
by a traditional Newick parser, such as ape::read.tree(). The function
read.raxml() can read such files and store the bootstrap as an additional
feature, which can be used to display on the tree or used to color tree
branches, etc.
raxml_file <- system.file("extdata/RAxML",
"RAxML_bipartitionsBranchLabels.H3",
package="treeio")
raxml <- read.raxml(raxml_file)
raxml## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/RAxML/RAxML_bipartitionsBranchLabels.H3'.
##
## ...@ phylo:
##
## Phylogenetic tree with 64 tips and 62 internal nodes.
##
## Tip labels:
## A_Hokkaido_M1_2014_H3N2_2014,
## A_Czech_Republic_1_2014_H3N2_2014,
## FJ532080_A_California_09_2008_H3N2_2008,
## EU199359_A_Pennsylvania_05_2007_H3N2_2007,
## EU857080_A_Hong_Kong_CUHK69904_2006_H3N2_2006,
## EU857082_A_Hong_Kong_CUHK7047_2005_H3N2_2005, ...
##
## Unrooted; includes branch lengths.
##
## with the following features available:
## 'bootstrap'.1.3.2.8 Parsing NHX tree
NHX (New Hampshire eXtended) format is an extension of Newick by introducing NHX tags. NHX is commonly used in phylogenetics software, including PHYLDOG (Boussau et al., 2013), RevBayes (Höhna et al., 2014), for storing statistical inferences. The following codes imported an NHX tree with associated data inferred by PHYLDOG (Figure 3.1A).
nhxfile <- system.file("extdata/NHX", "phyldog.nhx", package="treeio")
nhx <- read.nhx(nhxfile)
nhx## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/NHX/phyldog.nhx'.
##
## ...@ phylo:
##
## Phylogenetic tree with 16 tips and 15 internal nodes.
##
## Tip labels:
## Prayidae_D27SS7@2825365, Kephyes_ovata@2606431,
## Chuniphyes_multidentata@1277217,
## Apolemia_sp_@1353964, Bargmannia_amoena@263997,
## Bargmannia_elongata@946788, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'Ev', 'ND', 'S'.1.3.2.9 Parsing Phylip tree
Phylip format contains multiple sequence alignment of taxa in Phylip sequence
format with corresponding Newick tree text that was built from taxon sequences.
Multiple sequence alignment can be sorted based on the tree structure and displayed at
the right-hand side of the tree using ggtree through the msaplot() function or in combining with the ggmsa package (see also Basic Protocol 5 of (Yu, 2020)).
phyfile <- system.file("extdata", "sample.phy", package="treeio")
phylip <- read.phylip(phyfile)
phylip## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/sample.phy'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## K, N, D, L, J, G, ...
##
## Unrooted; no branch lengths.1.3.2.10 Parsing EPA and pplacer output
EPA (Berger et al., 2011) and PPLACER (Matsen et al., 2010) have a common output file format, jplace, which can be
parsed by the read.jplace() function.
jpf <- system.file("extdata/EPA.jplace", package="treeio")
jp <- read.jplace(jpf)
print(jp)## 'treedata' S4 object that stored information
## of
## '/home/ygc/R/library/treeio/extdata/EPA.jplace'.
##
## ...@ phylo:
##
## Phylogenetic tree with 493 tips and 492 internal nodes.
##
## Tip labels:
## CIR000447A, CIR000479, CIR000078, CIR000083,
## CIR000070, CIR000060, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'nplace'.The number of evolutionary placement on each branch will be calculated and stored as the nplace feature, which can be mapped to line size and/or color using ggtree (Yu et al., 2017).
1.3.2.11 Parsing jtree format
The jtree is a JSON-based format that was defined in
this treeio package (Wang et al., 2020) to support tree
data interchange (see session 3.3).
Phylogenetic tree with associated data can be exported to a single jtree
file using the write.jtree() function. The jtree can be easily parsed using any
JSON parser. The jtree format contains three keys: tree, data, and meta-data.
The tree value contains tree text extended from Newick tree format by putting
the edge number in curly braces after branch length. The data value contains
node/branch-specific data, while the meta-data value contains additional meta information.
jtree_file <- tempfile(fileext = '.jtree')
write.jtree(beast, file = jtree_file)
read.jtree(file = jtree_file)## 'treedata' S4 object that stored information
## of
## '/tmp/RtmpfSfWKq/file1aaef15aa0d8e.jtree'.
##
## ...@ phylo:
##
## Phylogenetic tree with 15 tips and 14 internal nodes.
##
## Tip labels:
## K_2013, N_2010, D_1987, L_1980, J_1983, G_1992, ...
##
## Rooted; includes branch lengths.
##
## with the following features available:
## 'height', 'height_0.95_HPD', 'height_range',
## 'length', 'length_0.95_HPD', 'length_median',
## 'length_range', 'rate', 'rate_0.95_HPD',
## 'rate_median', 'rate_range', 'height_median',
## 'posterior'.
1.3.3 Converting other tree-like objects to phylo or treedata objects
To extend the application scopes of treeio, tidytree and ggtree, treeio (Wang et al., 2020) provides several as.phylo and as.treedata methods to convert other tree-like objects, such as phylo4d and pml, to phylo or treedata object. So that users can easily map associated data to the tree structure, export a tree with/without data to a single file, manipulate and visualize a tree with/without data. These convert functions (Table 1.2) create the possibility of using tidytree to process tree using tidy interface and ggtree to visualize tree using the grammar of graphic syntax.
| Convert function | Supported object | Description |
|---|---|---|
| as.phylo | ggtree | convert ggtree object to phylo object |
| as.phylo | igraph | convert igraph object (only tree graph supported) to phylo object |
| as.phylo | phylo4 | convert phylo4 object to phylo object |
| as.phylo | pvclust | convert pvclust object to phylo object |
| as.phylo | treedata | convert treedata object to phylo object |
| as.treedata | ggtree | convert ggtree object to treedata object |
| as.treedata | phylo4 | convert phylo4 object to treedata object |
| as.treedata | phylo4d | convert phylo4d object to treedata object |
| as.treedata | pml | convert pml object to treedata object |
| as.treedata | pvclust | convert pvclust object to treedata object |
Here, we used pml object which was defined in the phangorn package, as an example. The pml() function computes the likelihood of a phylogenetic tree given a sequence alignment and a model and the optim.pml() function optimizes different model parameters. The output is a pml object, and it can be converted to a treedata object using as.treedata provided by treeio (Wang et al., 2020). The amino acid substitution (ancestral sequence estimated by pml) that stored in the treedata object can be visualized using ggtree as demonstrated in Figure 1.4.
library(phangorn)
treefile <- system.file("extdata", "pa.nwk", package="treeio")
tre <- read.tree(treefile)
tipseqfile <- system.file("extdata", "pa.fas", package="treeio")
tipseq <- read.phyDat(tipseqfile,format="fasta")
fit <- pml(tre, tipseq, k=4)
fit <- optim.pml(fit, optNni=FALSE, optBf=T, optQ=T,
optInv=T, optGamma=T, optEdge=TRUE,
optRooted=FALSE, model = "GTR",
control = pml.control(trace =0))
pmltree <- as.treedata(fit)
ggtree(pmltree) + geom_text(aes(x=branch, label=AA_subs, vjust=-.5))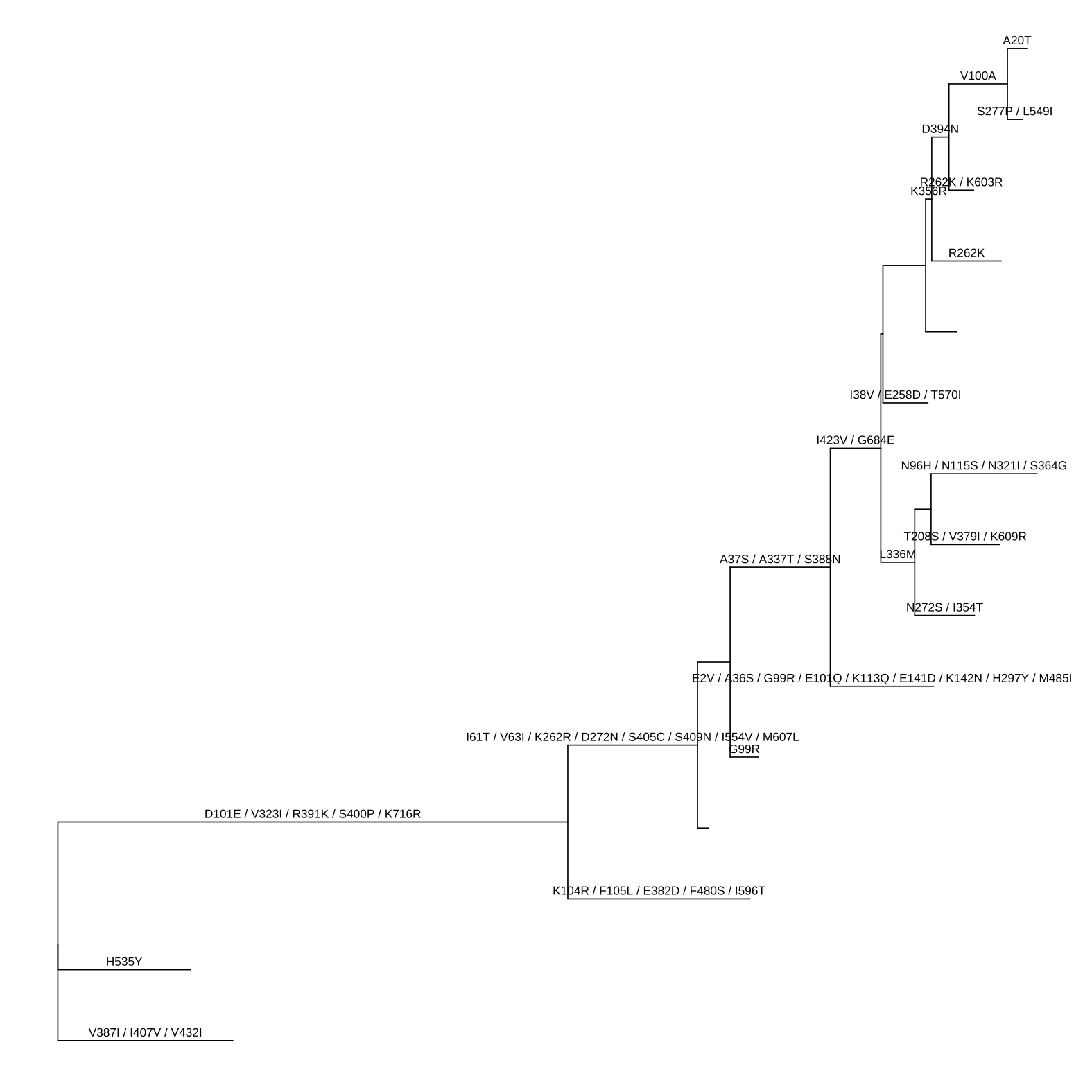
FIGURE 1.4: Converting pml object to treedata object. This allows using tidytree to process the tree data as well as using ggtree and ggtreeExtra to visualize the tree with associated data.
1.3.4 Getting information from treedata object
After the tree was imported, users may want to extract information stored
in the treedata object. treeio provides several accessor
methods to extract tree structure, features/attributes that stored in the object,
and their corresponding values.
The get.tree() or as.phylo() methods can convert the treedata object to a
phylo object which is the fundamental tree object in the R community and
many packages work with phylo object.
beast_file <- system.file("examples/MCC_FluA_H3.tree", package="ggtree")
beast_tree <- read.beast(beast_file)
# or get.tree
as.phylo(beast_tree)
beast_file <- system.file("examples/MCC_FluA_H3.tree", package="ggtree")
beast_tree <- read.beast(beast_file)
# or get.tree
print(as.phylo(beast_tree), printlen=3)##
## Phylogenetic tree with 76 tips and 75 internal nodes.
##
## Tip labels:
## A/Hokkaido/30-1-a/2013, A/New_York/334/2004, A/New_York/463/2005, ...
##
## Rooted; includes branch lengths.The get.fields method returns a vector of features/attributes stored in
the object and associated with the phylogeny.
get.fields(beast_tree)## [1] "height" "height_0.95_HPD"
## [3] "height_median" "height_range"
## [5] "length" "length_0.95_HPD"
## [7] "length_median" "length_range"
## [9] "posterior" "rate"
## [11] "rate_0.95_HPD" "rate_median"
## [13] "rate_range"The get.data method returns a tibble of all the associated data.
get.data(beast_tree)## # A tibble: 151 × 14
## height height_0.95_HPD height_median height_range
## <dbl> <list> <dbl> <list>
## 1 19 <dbl [2]> 19 <dbl [2]>
## 2 17 <dbl [2]> 17 <dbl [2]>
## 3 14 <dbl [2]> 14 <dbl [2]>
## 4 12 <dbl [2]> 12 <dbl [2]>
## 5 9 <dbl [2]> 9 <dbl [2]>
## 6 10 <dbl [2]> 10 <dbl [2]>
## 7 10 <dbl [2]> 10 <dbl [2]>
## 8 10.8 <dbl [2]> 10.8 <dbl [2]>
## 9 9 <dbl [2]> 9 <dbl [2]>
## 10 9 <dbl [2]> 9 <dbl [2]>
## # … with 141 more rows, and 10 more variables:
## # length <dbl>, length_0.95_HPD <list>,
## # length_median <dbl>, length_range <list>,
## # posterior <dbl>, rate <dbl>, rate_0.95_HPD <list>,
## # rate_median <dbl>, rate_range <list>, node <int>If users are only interested in a subset of the features/attributes returned by
get.fields, they can extract the information from the output of get.data or
directly subset the data by [ or [[.
beast_tree[, c("node", "height")]## # A tibble: 151 × 2
## node height
## <int> <dbl>
## 1 10 19
## 2 9 17
## 3 36 14
## 4 31 12
## 5 29 9
## 6 28 10
## 7 39 10
## 8 90 10.8
## 9 16 9
## 10 2 9
## # … with 141 more rows
head(beast_tree[["height_median"]])## height_median1 height_median2 height_median3
## 19 17 14
## height_median4 height_median5 height_median6
## 12 9 101.4 Summary
Software tools for inferring molecular evolution (e.g., ancestral states, molecular dating and selection pressure, etc.) are proliferating, but there is no single data format that is used by all different programs and capable to store different types of phylogenetic data. Most of the software packages have their unique output formats and these formats are not compatible with each other. Parsing software outputs is challenging, which restricts the joint analysis using different tools. The treeio package (Wang et al., 2020) provides a set of functions (Table 1.1) for parsing various types of phylogenetic data files and a set of converters (Table 1.2) to convert tree-like objects to phylo or treedata objects. These phylogenetic data can be integrated that allow further exploration and comparison. To date, most software tools in the field of molecular evolution are isolated and often not fully compatible with each other’s input and output files. These software tools are designed to do their analysis and the outputs are often not readable in other software. No tools have been designed to unify the inference data from different analysis programs. Efficient incorporation of data from different inference methods can enhance the comparison and understanding of the study target, which may help discover new systematic patterns and generate new hypotheses.
As phylogenetic trees are growing in their application to identify patterns in an evolutionary context, more different disciplines are employing phylogenetic trees in their research. For example, spatial ecologists may map the geographical positions of the organisms to their phylogenetic trees to understand the biogeography of the species (Schön et al., 2015); disease epidemiologists may incorporate the pathogen sampling time and locations into the phylogenetic analysis to infer the disease transmission dynamics in spatiotemporal space (Y.-Q. He et al., 2013); microbiologists may determine the pathogenicity of different pathogen strains and map them into their phylogenetic trees to identify the genetic determinants of the pathogenicity (Bosi et al., 2016); genomic scientists may use the phylogenetic trees to help taxonomically classify their metagenomic sequence data (Gupta & Sharma, 2015). A robust tool such as treeio to import and map different types of data into the phylogenetic tree is important to facilitate these phylogenetics-related research, or ‘phylodynamics’. Such tools could also help integrate different meta-data (time, geography, genotype, epidemiological information) and analysis results (selective pressure, evolutionary rates) at the highest level and provide a comprehensive understanding of the study organisms. In the field of influenza research, there have been such attempts of studying the phylodynamics of the influenza virus by mapping different meta-data and analysis results on the same phylogenetic tree and evolutionary timescale (Lam et al., 2015).