7 Differential Expression & Pseudobulk
Finding marker genes is the bread and butter of single-cell analysis. But single-cell Differential Expression (DE) is notoriously tricky. Due to the vast number of cells (which inflates statistical power) and the zero-inflated nature of the data, standard single-cell DE often yields an overwhelming number of “significant” genes that turn out to be false positives or artifacts of biological replicates.
Reviewers at high-impact journals (like Nature Methods) are increasingly demanding Pseudobulk analysis to control for false discovery rates. sclet makes both standard DE and Pseudobulk DE incredibly straightforward.
In this chapter, the standard single-cell DE examples are executed during book rendering so they also serve as lightweight regression checks. The pseudobulk section now includes a small CI-friendly smoke test built from synthetic donor groupings, while the surrounding discussion still reflects the fact that real pseudobulk designs should be driven by actual biological replicates.
7.1 Standard Single-Cell DE
You can find markers for all clusters using RunDEtest(), which wraps FindMarkers or FindAllMarkers. Importantly, RunDEtest() saves the result as an analysis-state inside the SingleCellExperiment object.
library(sclet)
# Assuming 'sce' has been processed and clustered
sce <- readRDS("data/pbmc-3k-sce.rds")
# Run differential expression test and record the state
sce <- RunDEtest(sce, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
# Visualize the DE results using a Volcano plot
DEtestPlot(sce)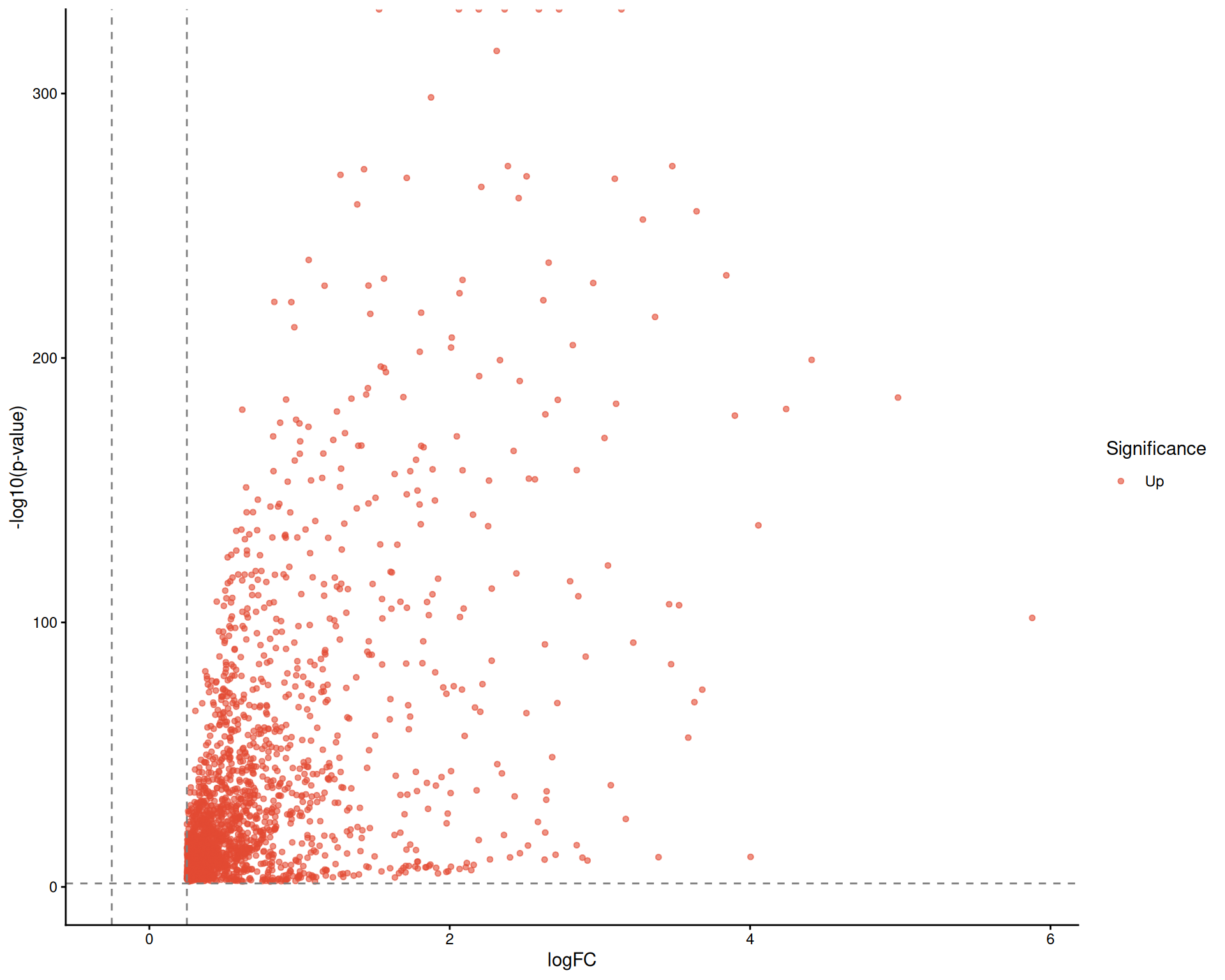
7.1.1 The “What does this gene even do?” Problem
When you get a list of marker genes, you’re often left staring at cryptic acronyms like ZBTB16 or KLRB1. What do they actually do?
sclet provides a handy utility called gene_summary_table() that automatically fetches the full name and biological summary of the genes from the selected orgDb annotation package.
# Get the raw DE results from the state
de_results <- get_detest(sce)$results
# Take the top 5 markers
top_genes <- head(de_results$gene, 5)
# Fetch their biological summaries!
summary_df <- gene_summary_table(
top_genes,
orgDb = "org.Hs.eg.db",
output = "data.frame"
)
print(summary_df)## [1] ENTREZID gene name description
## [5] summary
## <0 rows> (or 0-length row.names)This simple helper can save you hours of manually Googling gene names.
7.2 The Gold Standard: Pseudobulk Analysis
To avoid the inflated p-values of single-cell DE, the current best practice is to aggregate the counts of all cells within the same biological replicate and cell type, creating a “pseudobulk” sample. You then run robust bulk RNA-seq tools (like DESeq2 or edgeR) on these aggregated samples.
sclet handles this elegantly with two functions: AggregateExpression() and FindMarkers_pseudobulk().
7.2.1 Step 1: Aggregate Expression
In a real analysis, your sce object would usually already contain replicate metadata such as donor_id and cell_type. For a runnable book example, we create a simple donor column on the fly and reuse the current cluster labels as grouping labels.
# For a runnable example, we create a simple donor column.
cluster_id <- as.character(SingleCellExperiment::colLabels(sce))
if (length(cluster_id) == 0) {
cluster_id <- as.character(Idents(sce))
}
sce$cluster_id <- cluster_id
sce$donor_id <- rep(paste0("donor", 1:4), length.out = ncol(sce))
# Aggregate counts by cluster and donor
pb_sce <- AggregateExpression(
sce,
group_by = c("cluster_id", "donor_id"),
assay.type = "counts"
)
# The result is a much smaller SummarizedExperiment
# where each column is a "donor-cluster" pseudobulk sample.
dim(pb_sce)## [1] 13714 407.2.2 Step 2: Pseudobulk DE with DESeq2
Now, you can run DESeq2 directly on this aggregated object to find markers robustly.
cluster_tab <- table(pb_sce$cluster_id)
target_clusters <- names(cluster_tab[cluster_tab >= 2])[1:2]
stopifnot(length(target_clusters) == 2)
pb_subset <- pb_sce[, pb_sce$cluster_id %in% target_clusters]
res <- FindMarkers_pseudobulk(
pb_subset,
design = ~ cluster_id,
contrast = c("cluster_id", target_clusters[1], target_clusters[2])
)
head(res[, c("log2FoldChange", "pvalue", "padj")])By leveraging AggregateExpression() and FindMarkers_pseudobulk(), you completely bypass the inflated p-value problem and ensure your biological findings are robust across actual replicates, not just individual cells.