ggmsa: Getting Started
2021-09-22
ggmsa.RmdIntroduction
ggmsa is a package designed to plot multiple sequence alignments。
This package implements functions to visualize publication-quality multiple sequence alignments (protein/DNA/RNA) in R extremely simple and powerful. It uses module design to annotate sequence alignments and allows to accept other datasets for diagrams combination.
In this tutorial, we’ll work through the basics of using ggmsa.
library("ggmsa")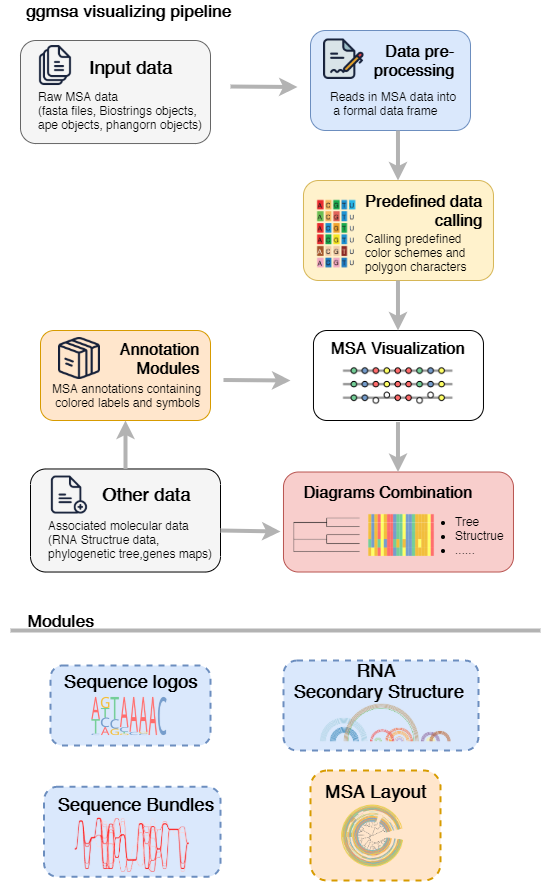
Importing MSA data
We’ll start by importing some example data to use throughout this tutorial. Expect FASTA files, some of the objects in R can also as input. available_msa() can be used to list MSA objects currently available.
available_msa()
#> files currently available:
#> .fasta
#> XStringSet objects from 'Biostrings' package:
#> DNAStringSet RNAStringSet AAStringSet BStringSet DNAMultipleAlignment RNAMultipleAlignment AAMultipleAlignment
#> bin objects:
#> DNAbin AAbin
protein_sequences <- system.file("extdata", "sample.fasta", package = "ggmsa")
miRNA_sequences <- system.file("extdata", "seedSample.fa", package = "ggmsa")
nt_sequences <- system.file("extdata", "LeaderRepeat_All.fa", package = "ggmsa")
Basic use: MSA Visualization
The most simple code to use ggmsa:
ggmsa(protein_sequences, 300, 350, color = "Clustal", font = "DroidSansMono", char_width = 0.5, seq_name = TRUE )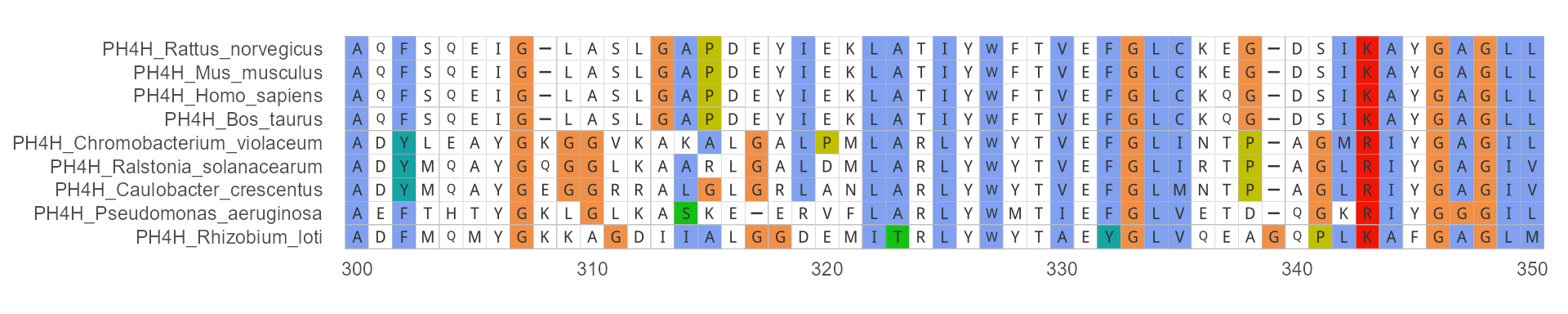
Color Schemes
ggmsa predefines several color schemes for rendering MSA are shipped in the package. In the same ways, using available_msa() to list color schemes currently available. Note that amino acids (protein) and nucleotides (DNA/RNA) have different names.
available_colors()
#> color schemes for nucleotide sequences currently available:
#> Chemistry_NT Shapely_NT Taylor_NT Zappo_NT
#> color schemes for AA sequences currently available:
#> Clustal Chemistry_AA Shapely_AA Zappo_AA Taylor_AA LETTER CN6 Hydrophobicity
Font
Several predefined fonts are shipped ggmsa. Users can use available_fonts() to list the font currently available.
available_fonts()
#> font families currently available:
#> helvetical mono TimesNewRoman DroidSansMonoMSA Annotation
ggmsa supports annotations for MSA. Similar to the ggplot2, it implements annotations by geom and users can perform annotation with + , like this: ggmsa() + geom_*(). Automatically generated annotations that containing colored labels and symbols are overlaid on MSAs to indicate potentially conserved or divergent regions.
For example, visualizing multiple sequence alignment with sequence logo and bar chart:
ggmsa(protein_sequences, 221, 280, seq_name = TRUE, char_width = 0.5) + geom_seqlogo(color = "Chemistry_AA") + geom_msaBar()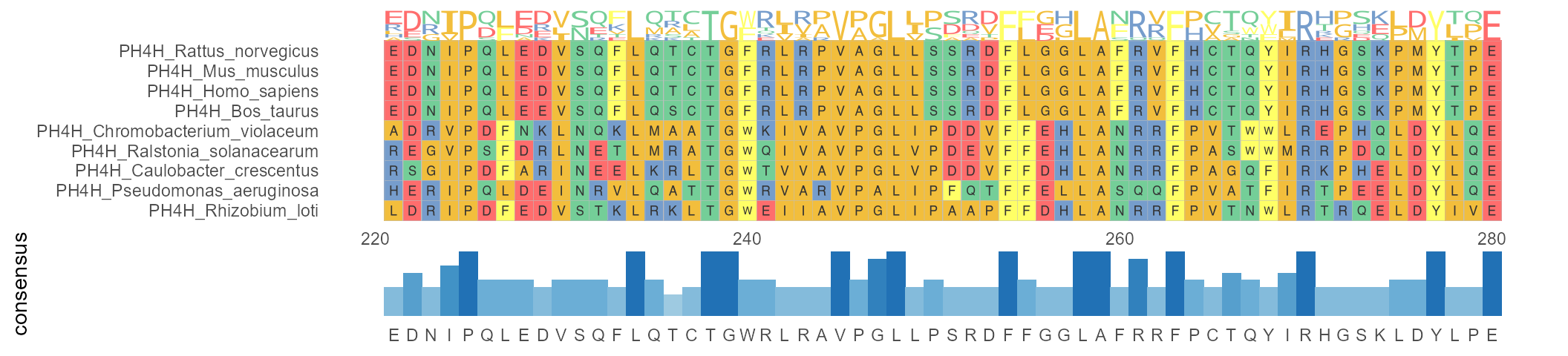
This table shows the annnotation layers supported by ggmsa as following:
| Annotation modules | Type | Description |
|---|---|---|
| geom_seqlogo() | geometric layer | automatically generated sequence logos for a MSA |
| geom_GC() | annotation module | shows GC content with bubble chart |
| geom_seed() | annotation module | highlights seed region on miRNA sequences |
| geom_msaBar() | annotation module | shows sequences conservation by a bar chart |
| geom_helix() | annotation module | depicts RNA secondary structure as arc diagrams(need extra data) |