The clusterProfiler package provides the bitr() and bitr_kegg() functions for converting ID types. Both bitr() and bitr_kegg() support many species including model and many non-model organisms.
For GO analysis, users don’t need to convert IDs, as all ID types provided by OrgDb can be used in groupGO, enrichGO, and gseGO by specifying the keyType parameter.
Users can use the bitr function to convert IDs using any ID types available in the OrgDb object. For example, users may want to know the genes in the background that belong to a specific GO term. Such information can be easily accessed using bitr.
m <-bitr("GO:0006805", fromType="GO", toType ="SYMBOL", OrgDb=org.Hs.eg.db)dim(m)
[1] 168 4
head(m)
GO EVIDENCE ONTOLOGY SYMBOL
1 GO:0006805 TAS BP NAT1
2 GO:0006805 TAS BP NAT2
3 GO:0006805 TAS BP AADAC
4 GO:0006805 TAS BP ADA
5 GO:0006805 IEA BP AHR
6 GO:0006805 TAS BP AHR
Note: If you want to extract genes in your input gene list that are belong to a specific term/pathway, you can use the geneInCategory function.
19.1.2bitr_kegg: converting biological IDs using KEGG API
The bitr_kegg() function supports ID conversion via the KEGG API, which is useful for species supported by KEGG but lacking an OrgDb object. For detailed usage and examples, please refer to the KEGG ID Conversion section in the KEGG analysis chapter.
19.2setReadable: translating gene IDs to human readable symbols
Some of the functions, especially those internally supported for DO, GO, and Reactome Pathway, support a parameter, readable. If readable = TRUE, all the gene IDs will be translated to gene symbols. The readable parameter is not available for enrichment analysis of KEGG or using user’s own annotation. KEGG analysis using enrichKEGG and gseKEGG, which internally query annotation information from KEEGG database and thus support all species if it is available in the KEGG database. However, KEGG database doesn’t provide gene ID to symbol mapping information. For analysis using user’s own annotation data, we even don’t know what species is in analyzed. Translating gene IDs to gene symbols is partly supported using the setReadable function if and only if there is an OrgDb available. The setReadable() function also works with compareCluster() output.
data(geneList, package="DOSE")de <-names(geneList)[1:100]x <-enrichKEGG(de)## The geneID column is ENTREZIDhead(x, 3)
For those functions that internally support readable parameter, user can also use setReadable for translating gene IDs.
19.3 Parsing GMT files
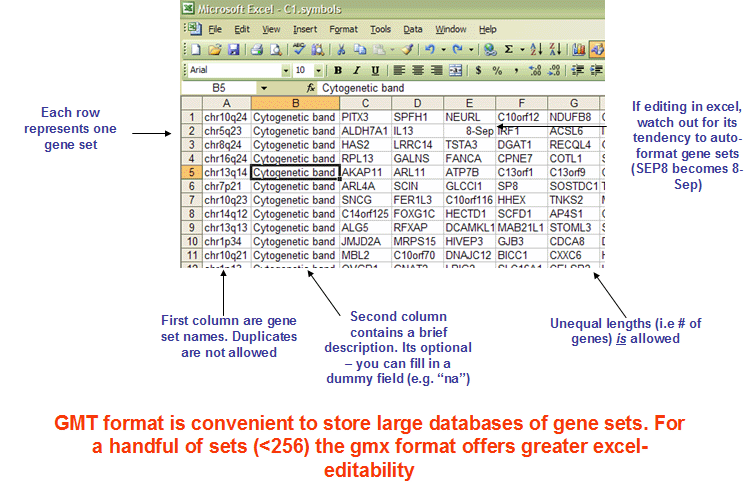
The GMT (Gene Matrix Transposed) file format is a tab delimited file format that is widely used to describe gene sets. Each row in the GMT format represents one gene set and each gene set is described by a name (or ID), a description and the genes in the gene set as illustrated in Figure 19.1.
The clusterProfiler package implemented a function, read.gmt(), to parse GMT file into a data.frame. The WikiPathway GMT file encodes information of version, wpid and species into the Description column. The clusterProfiler provides the read.gmt.wp() function to parse WikiPathway GMT file and supports parsing information that encoded in the Description column.
# use `wget -c` in `download.file`wget::wget_set()url <-"https://maayanlab.cloud/Enrichr/geneSetLibrary?mode=text&libraryName=COVID-19_Related_Gene_Sets"download.file(url, destfile ="COVID19_GeneSets.gmt")covid19_gs <-read.gmt("COVID19_GeneSets.gmt")head(covid19_gs)
term gene
1 COVID19-E protein host PPI from Krogan BRD4
2 COVID19-E protein host PPI from Krogan BRD2
3 COVID19-E protein host PPI from Krogan SLC44A2
4 COVID19-E protein host PPI from Krogan ZC3H18
5 COVID19-E protein host PPI from Krogan AP3B1
6 COVID19-E protein host PPI from Krogan CWC27
There are many gene sets available online. After parsing by the read.gmt() function, the data can be directly used to perform enrichment analysis using enricher() and GSEA() functions (see also Chapter 12).
19.4 Data frame interface for accessing enriched results
Enrichment result objects in clusterProfiler support standard data frame operations including [, [[, and $ operators, allowing users to subset and access results using familiar data frame syntax.
19.4.1 Subsetting with [ operator
The [ operator can be used to subset enrichment results based on row indices or logical conditions:
# Subset first 5 rowsx_first5 <- x[1:5, ]# Subset based on p-value thresholdx_sig <- x[x$pvalue <0.05, ]# Subset specific terms by IDx_specific <- x[x$ID %in%c("hsa04110", "hsa04218"), ]
By default, the [ operator returns a data.frame. To preserve the enrichment result object structure for further analysis and visualization, use the asis = TRUE parameter:
# Preserve as enrichResult objectx_filtered <- x[x$pvalue <0.05, asis =TRUE]class(x_filtered)
Users can filter enrichment results based on various criteria:
# Filter by adjusted p-valuex_padj <- x[x$p.adjust <0.05, asis =TRUE]# Filter by gene count (minimum 5 genes per term)x_min5 <- x[x$Count >=5, asis =TRUE]# Filter specific biological processesx_bp <- x[grepl("process", x$Description, ignore.case =TRUE), asis =TRUE]# Filter terms containing specific genesx_containing_gene <- x[grepl("6280", x$geneID), asis =TRUE]
This data frame interface provides flexible filtering capabilities while maintaining compatibility with enrichplot visualization functions when using asis = TRUE.
19.5 Alternative filtering approaches with dplyr
While base R operators provide familiar syntax for filtering enrichment results, users may prefer the more readable and chainable approach offered by dplyr verbs. For comprehensive examples of using filter, arrange, select, mutate, and other dplyr functions with enrichment result objects, please refer to the dplyr verbs for manipulating enrichment result chapter.
The dplyr approach is particularly useful for complex filtering operations and pipeline-style data manipulation.