Semantic Knowledge Mining: Methods and Applications
We have developed methods and tools for biological knowledge mining, including: (1) semantic similarity measurement; (2) universal enrichment analysis; (3) comparative analysis of biological themes; (4) cistrome data analysis; (5) single-cell data analysis; and (6) spatial transcriptomics analysis. These methods and software tools support the application of biomedical knowledge across diverse species, facilitating the mining of biological data and the discovery of novel insights.
1. Semantic Similarity Measurement Link to heading
| Gene functional similarity calculations mathematically quantify biomedical knowledge to gauge the similarity between genes. This forms the basis for solving various challenges, such as predicting gene attributes and analyzing diseases and drugs. To facilitate this process, we developed the software tool GOSemSim, built upon gene ontology principles. This work was published in Bioinformatics in 2010. We have extended this work to encompass diverse biomedical knowledge domains, including diseases, phenotypes, and medical subject headings. |
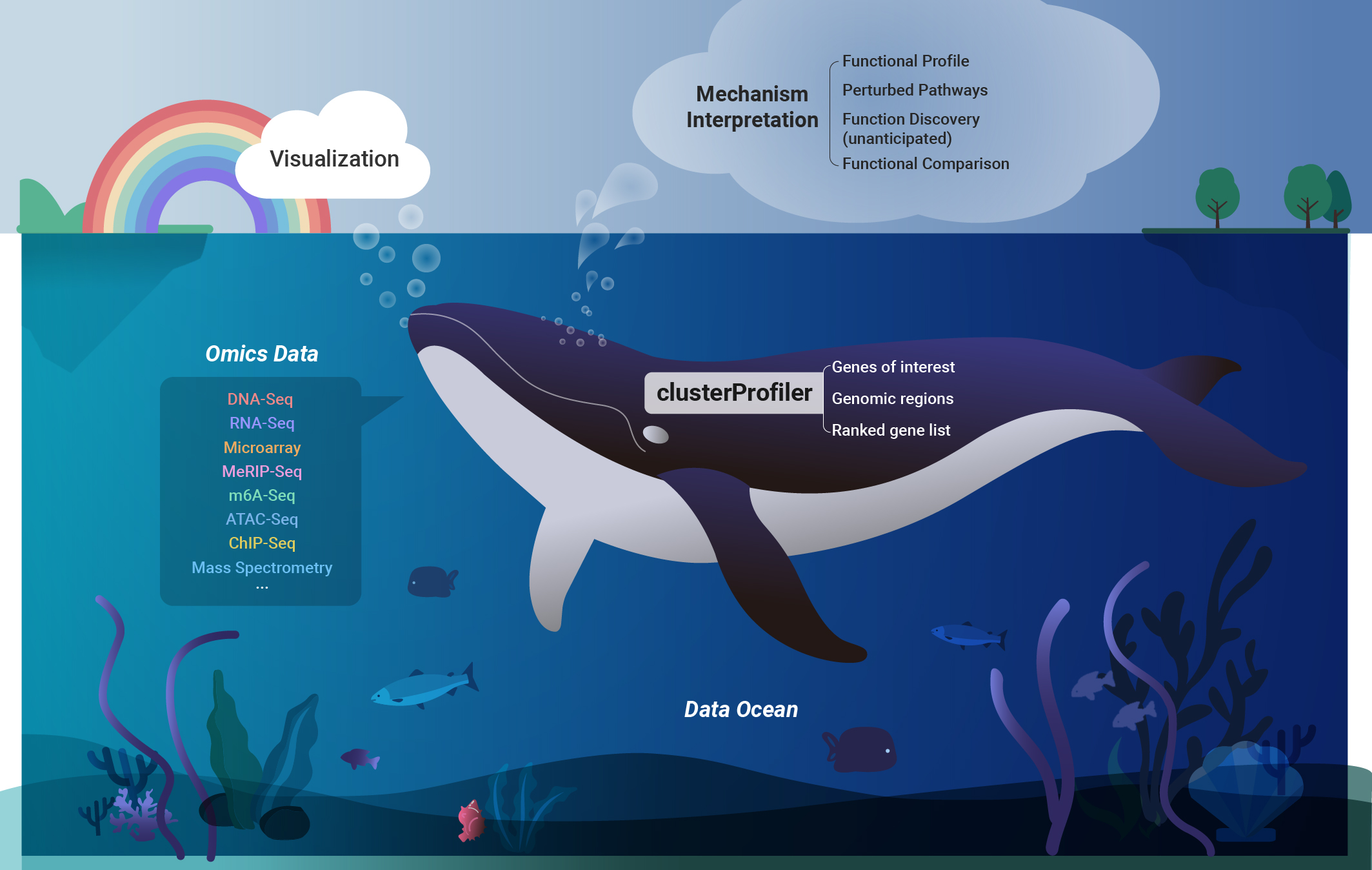
2. Functional Enrichment Analysis Link to heading
| To identify biological pathways associated with perturbed genes, we developed a suite of R packages, spearheaded by clusterProfiler. This tool facilitates online retrieval of genome annotations, supporting enrichment analysis for thousands of species. It also provides a universal interface for user-provided custom annotations. Notably, clusterProfiler supports the utilization of genomic regions for enrichment analysis. We have extended its application to disease ontology, medical subject headings, and Reactome pathway analysis. This work has been widely cited and integrated into numerous bioinformatics software tools. |
3. Comparative Biological Theme Analysis Link to heading
| To address complex experimental designs involving multiple time points and conditions, we developed clusterProfiler for comparing biological themes. This contribution was published in OMICS: A Journal of Integrative Biology in 2012. The tool enables users to specify complex experimental designs using formula syntax, allowing for the comparison of functional profiles across different conditions. We authored a protocol article in Nature Protocols in 2024, detailing the application of this comparative framework. |

4. Cistrome Data Analysis Link to heading
| We developed the ChIPseeker package to bridge the gap between genomic cis-regulatory positions and biological function. It facilitates annotation and comparison of genomic peaks, integrating with the GEO database for data mining. This work was published in Bioinformatics in 2015. ChIPseeker is widely used for analyzing various cistromic datasets, including ChIP-seq, DNase-seq, and ATAC-seq. |
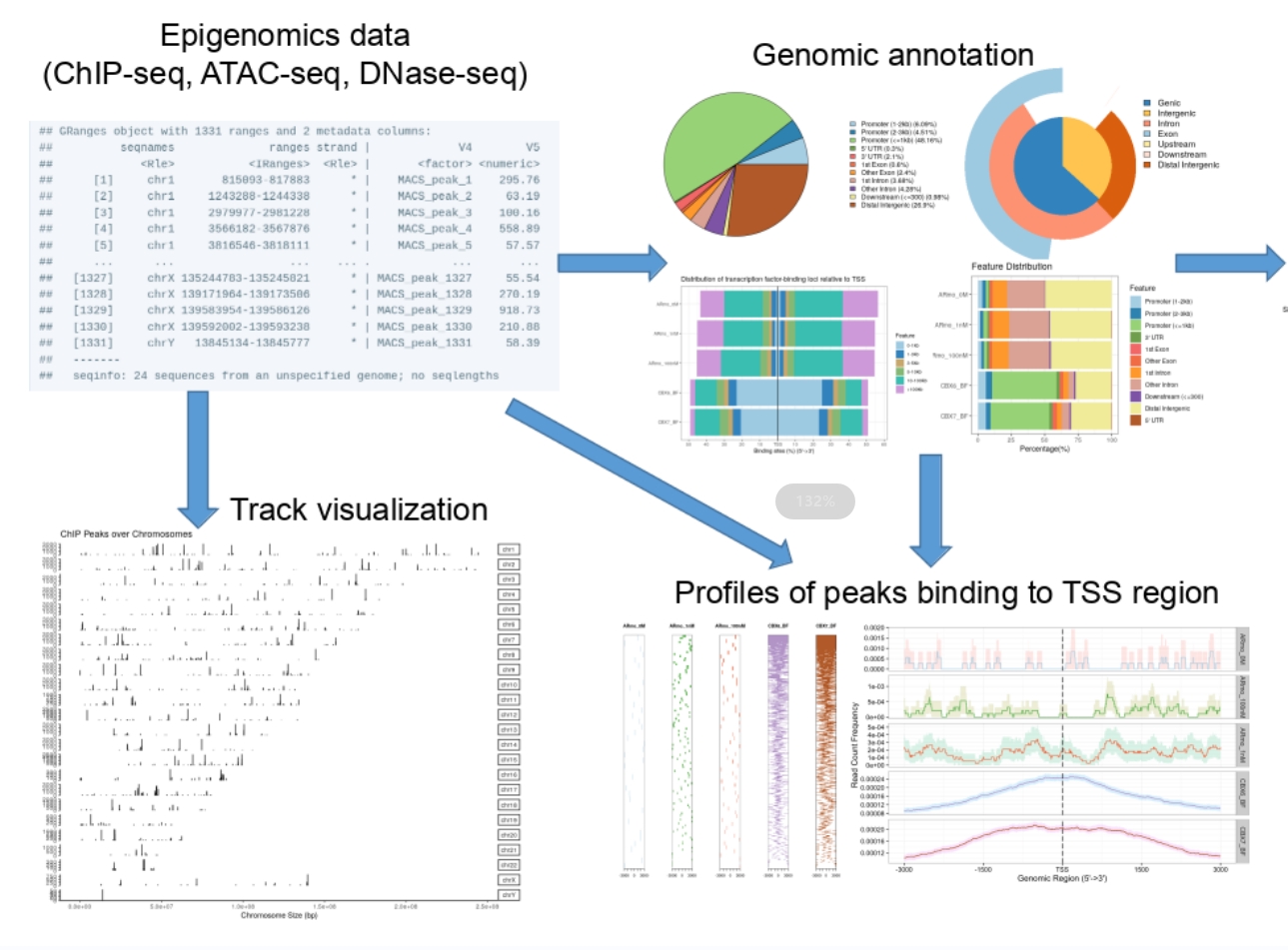
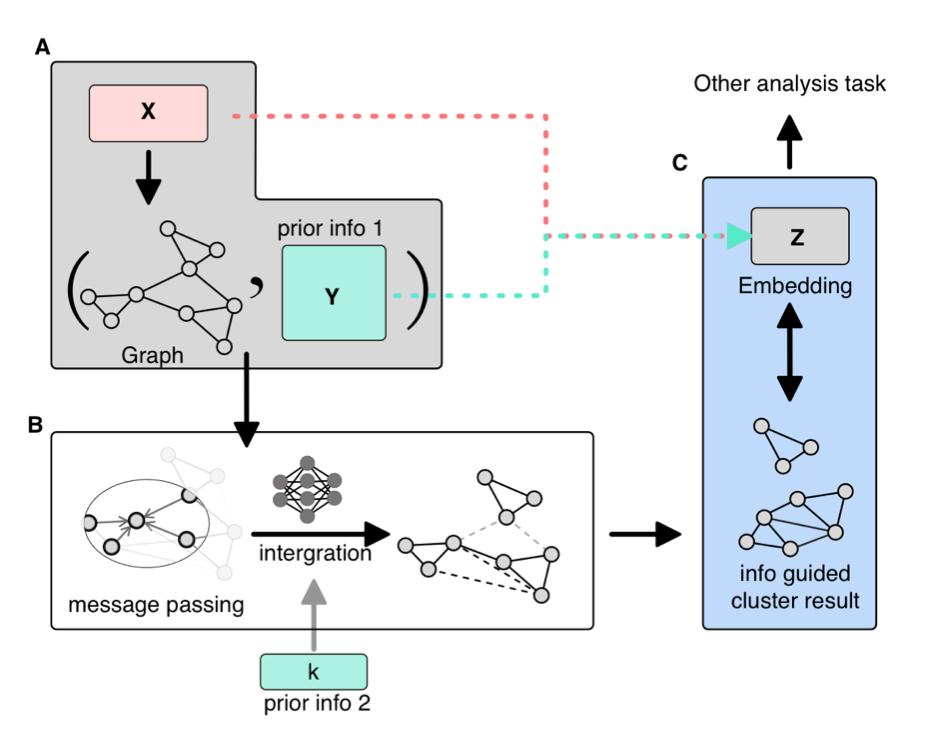
5. Single-Cell Data Analysis Link to heading
| To enhance the interpretability of single-cell clustering, we proposed integrating biological knowledge as attributes of graph nodes. We developed the MSGNN tool based on graph neural networks to realize this advancement, improving the biological interpretation of complex data. |
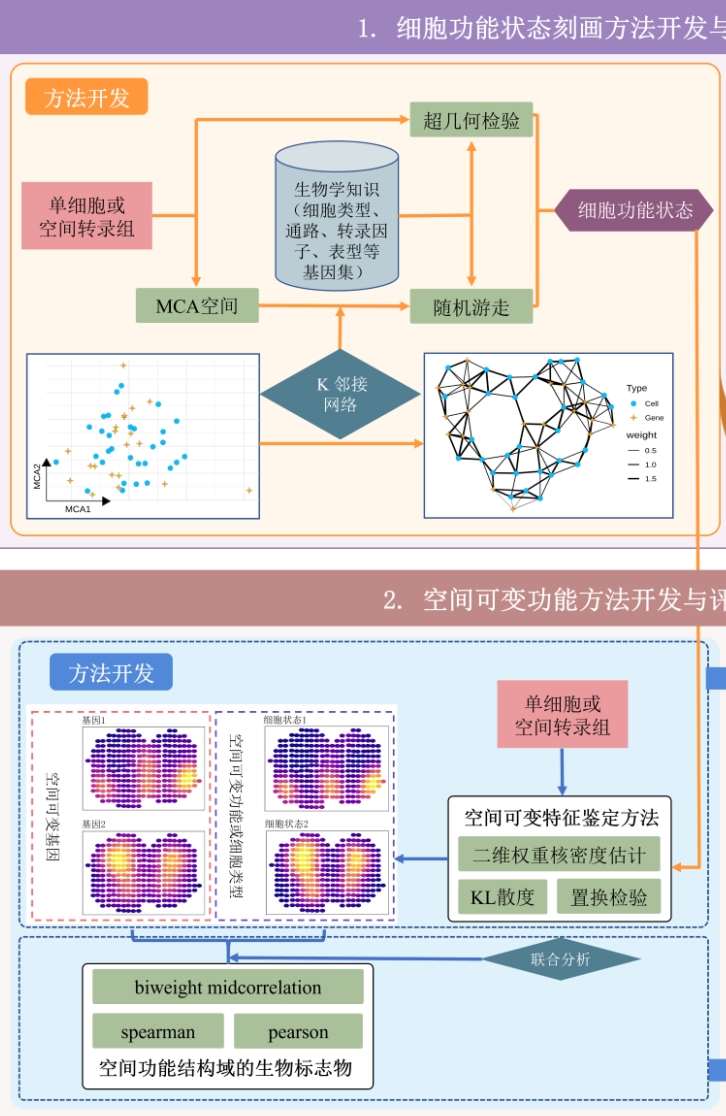
6. Spatial Transcriptomics Analysis Link to heading
| We proposed a method for characterizing cell functional states and identifying spatially highly variable features. We developed the SVP package to facilitate these methods, enabling the characterization of biological functions and the determination of their spatial distribution specificity. |
Feedback from the academic community Link to heading
|
Publications Link to heading
- G Yu*. Background bias in functional enrichment analysis: Insights from clusterProfiler. The Innovation Life. 2026, 4(1):100181.
- G Yu*. Thirteen years of clusterProfiler. The Innovation. 2024, 5(6):100722.
- S Xu#, E Hu#, Y Cai#, Z Xie#, X Luo#, L Zhan, W Tang, Q Wang, B Liu, R Wang, W Xie, T Wu, L Xie, G Yu*. Using clusterProfiler to characterise Multi-Omics Data. Nature Protocols. 2024, 19(11):3292-3320.
- Q Wang#, M Li#, T Wu, L Zhan, L Li, M Chen, W Xie, Z Xie, E Hu, S Xu, G Yu*. Exploring epigenomic datasets by ChIPseeker. Current Protocols, 2022, 2(10): e585.
- N Sato, Y Tamada, G Yu, Y Okuno*. CBNplot: Bayesian network plots for enrichment analysis. Bioinformatics. 2022, 38(10):2959-2960.
- T Wu#, E Hu#, S Xu, M Chen, P Guo, Z Dai, T Feng, L Zhou, W Tang, L Zhan, X Fu, S Liu, X Bo*, G Yu*. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021, 2(3):100141.
- Yu G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. In: Kidder B. (eds) Stem Cell Transcriptional Networks. Methods in Molecular Biology, 2020, 2117:207-215. Humana, New York, NY.
- Z Hao, D Lv, Y Ge, J Shi, D Weijers, G Yu*, J Chen*. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Computer Science. 2020, 6:e251.
- G Yu. Using meshes for MeSH term enrichment and semantic analyses. Bioinformatics. 2018, 34(21):3766-3767.
- G Yu, QY He*. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Molecular BioSystems. 2016, 12(2):477-479.
- G Yu*, LG Wang, QY He*. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparision and visualization. Bioinformatics. 2015, 31(14):2382-2383.
- G Yu*, LG Wang, GR Yan, QY He*. DOSE: an R/Bioconductor package for Disease Ontology Semantic and Enrichment analysis. Bioinformatics. 2015, 31(4):608-609.
- G Yu, LG Wang, Y Han, QY He*. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.
- G Yu, QY He*. Functional similarity analysis of human virus-encoded miRNAs. Journal of Clinical Bioinformatics, 2011, 1(1):15.
- G Yu#, CL Xiao#, X Bo, CH Lu, Y Qin, S Zhan, QY He*. A new method for measuring functional similarity of microRNAs. Journal of Integrated OMICS, 2011, 1(1):49-54.
- G Yu#, F Li#, Y Qin, X Bo*, Y Wu, S Wang*. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010, 26(7):976-978.