Exploring Biological Knowledge and Discovery
Knowledge discovery within precision medicine big data is crucial for advancing clinical translational applications. By leveraging biomedical knowledge, we can facilitate the uncovering of new insights in biomedicine. We have developed a suite of methods and tools, including: (1) pioneering biological theme comparison for complex experimental designs, (2) universal enrichment analysis methods for omics data interpretation, (3) semantic similarity measurement to aid in biological knowledge discovery, (4) cistromic data mining for identifying co-regulators, (5) integration of biological knowledge to enhance single-cell clustering interpretability, and (6) characterization of single-cell functional states and identification of spatial specific biological functions. These methods and software broaden the application of biomedical knowledge across diverse species, facilitating biological big data mining and uncovering novel disoveries.
1. Frist-time implementation of comparative biological theme analysis Link to heading
|
|
As experimental designs evolved beyond simple case-control setups, encompassing complex configurations involving multiple time points and conditions became imperative for studying biomedical phenomena. Recognizing this need, We devloped the clusterProfiler for comparing biological themes, a pivotal contribution published in OMICS: A Journal of Integrative Biology in 2012. This work has garnered over 20,000 citations, making it the most highly cited paper in the journal and one of China’s top 10 highly cited papers from 2011 to 2021. With its exceptionally user-friendly interface, clusterProfiler enables users to specify complex experimental designs using formula syntax. This capability as exemplified in an article published in The Innovation in 2021, where data showcasing different time points with various drug treatments simultaneously were presented. Twelve years after its initial publication, we authored a protocol-type article introducing the functionality of comparing biological themes in disease subtypes, functions of perturbed transcription factors, and cell type annotations using various omics data sets, including metagenomics, metabolomics, transcriptomics, and single-cell omics. This work was published in Nature Protocols in 2024 and was selected as a feature protocol of the journal. |

2. Uncovering potential molecular mechanisms through high-throughput data and biomedical knowledge Link to heading
|
|
One pivotal aspect of functional genomics research is the identification the biological pathways associated with perturbed genes and the formulation of hypotheses regarding molecular mechanisms. While gene enrichment analysis serves as a widely adopted and effective method, it encounters challenges such as outdated annotation and limited supports for non-model organisms. To address these issues, we have developed a suite of R packages, spearheaded by clusterProfiler. ClusterProfiler offers several key features to overcome these challenges. It facilitates online retrieval of the latest genome annotations, supporting Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for thousands of species. Additionally, it provides a universal interface for incorporating user-provided custom annotations, enabling analysis of new species and utilization of new functional annotations. Notably, clusterProfiler allows for the utilization of genomic regions, enabling enrichment analysis of epigenomic data. Moreover, it implements comparative analysis of multiple datasets to support complex experimental designs and offers a tidy interface, streamlining data manipulation and interpretation. Published in OMICS: A Journal of Integrative Biology in 2012, The Innovation in 2021 and Nature Protocols in 2024, this work has made a substantial impact on biomedical research, accumulating over 30,000 citations and being integrated into more than 40 bioinformatics software tools. It has emerged as an indispensable tool for various omics data analyses. Furthermore, we have extended its application to disease ontology (Bioinformatics 2015), medical subject headings (Bioinformatics 2018), and Reactome pathway analysis (Molecular BioSystems 2016), further expanding its utility in biomedical research. |
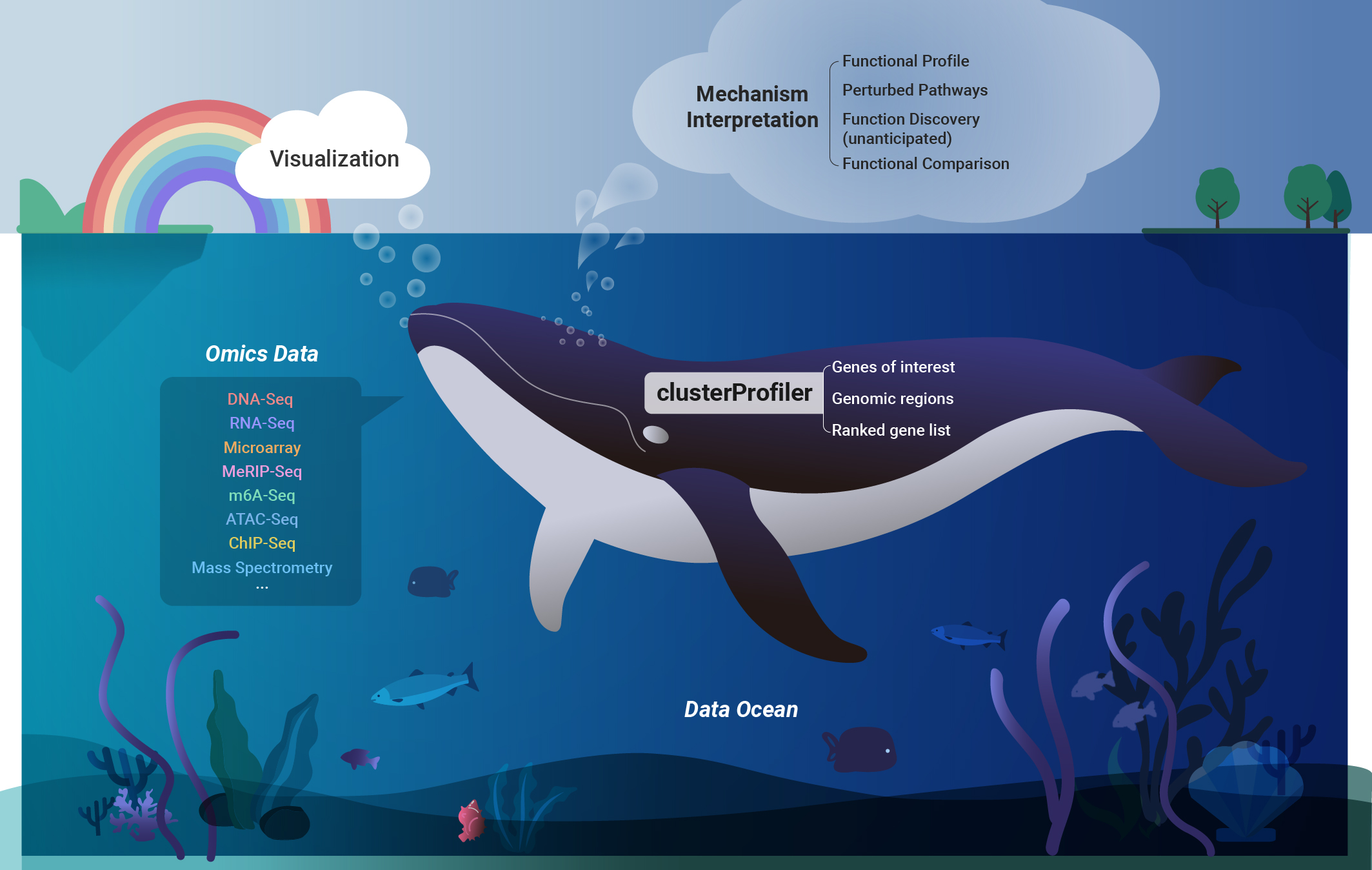
3. Biological knowledge discovery through gene semantic similarity measurement Link to heading
|
|
By leveraging biomedical knowledge, novel insights can be unearthed within the field. Gene functional similarity calculations, rooted in biomedical knowledge, play a pivotal role in this endeavor. These calculations methematically quantify biomedical knowledge to gauge the similarity between genes. Similarity entities share similar functions and behaviors, which form the basis for solving a wide range of real-world challenges. These challenges span predicting and inferring gene attributes such as function, localization, and interactions, as well as analyzing diseases and drugs, including drug repurposing. To facilitate this process, we developed the software tool GOSemSim, built upon gene ontology principles. GOSemSim incorporates multiple information content-based algorithms and graph-based algorithms to provide comprehensive gene similarity assessments. This work was initially published in Bioinformatics in 2010 and later featured as a chapter in Stem Cell Transcriptional Networks (2nd edition) in 2020. Subsequently, we extended the scope of this work to encompass a broader array of biomedical knowledge domains. This expanded version supports the measurement of gene similarity from diverse perspectives, including diseases (Bioinformatics 2015), phenotypes, and medical subject headings (Bioinformatics 2018). |
4. Annotating, visualizing, mining of cistrome data Link to heading
|
|
The term “cistrome” refers to the ensemble of cis-acting targets of trans-acting factors across the genome, encompassing transcription factor binding sites and the genomic loci of histone modifications. Genomic cis-regulatory information is typically obtained on a genome-wide scale through techniques such as ChIP-seq, DNase-seq, and ATAC-seq. Bridging the gap between upstream (locating sites) and downstream analysis (functional investigations) hinges on annotating the genomic positional data effectively. However, existing tools are often tailored for individual datasets, posing a challenge as cistromic technologies proliferate and public data accumulates. Comparing multiple datasets, mining public data, and identifying potential co-regulators of transcription factors are essential for comprehensive understanding of cistrome interactions. To tackle these challenges, we developed the ChIPseeker package for annotation and comparision, seamlessly integrated with the GEO database to facilitate data mining for potential co-transcription factors or protein complexes. This pivotal work was published in Bioinformatics in 2015. ChIPseeker has since become a cornerstone in the analysis of DNase-seq and ATAC-seq data, widely employed across various cistromic datasets. Additionally, we published a protocol article in Current Protocols in 2022, detailing the application of ChIPseeker in analyzing diverse epigenomic datasets, further extending its utility and impact in the field. |
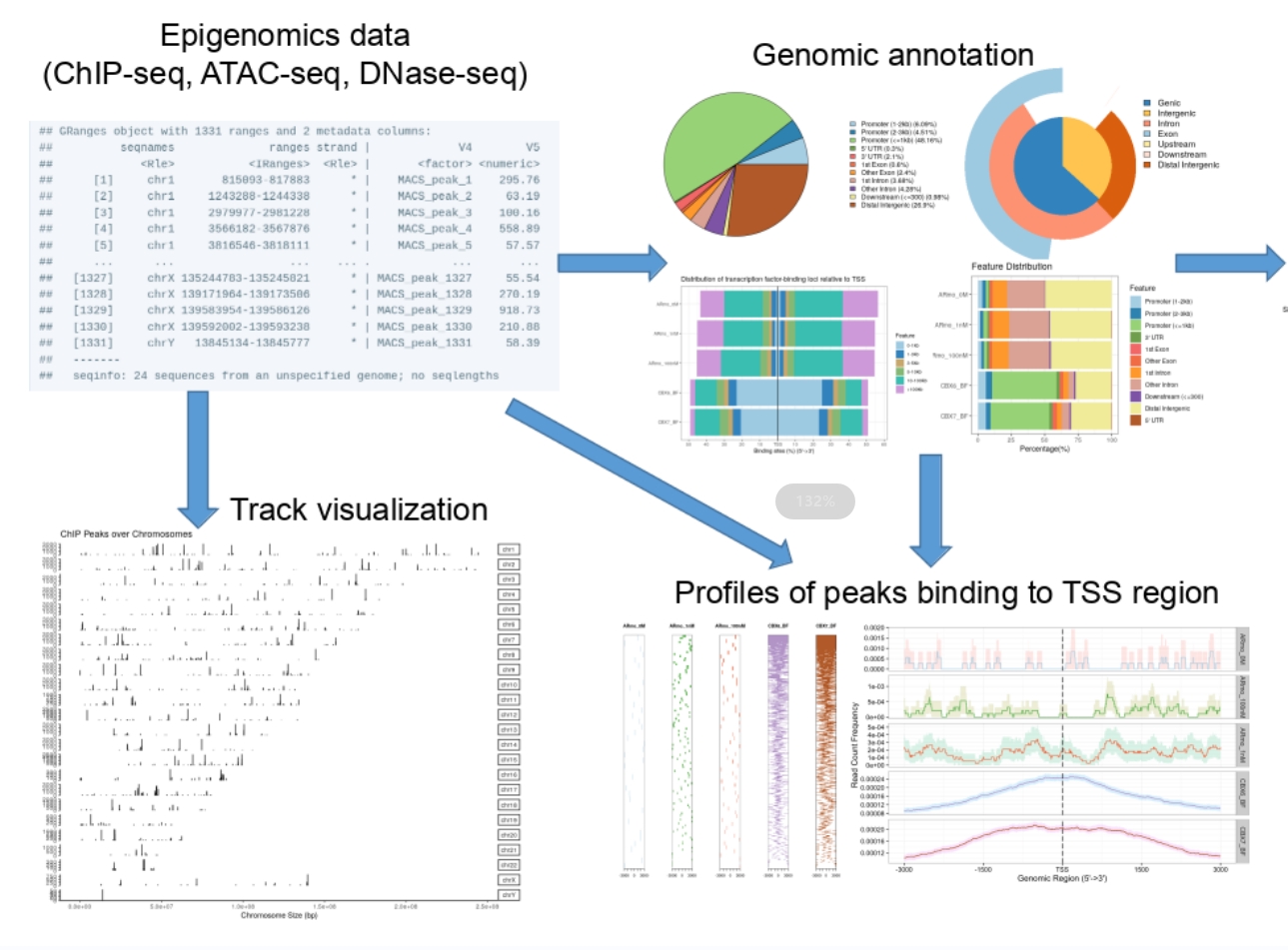
5. Enhancing single-cell clustering interpretability through biological knowledge integration Link to heading
|
|
Clustering serves as a pivotal bridge between upstream and downstream analyses in single-cell analysis, with precise clustering being essential for subsequent investigations. Presently, prevalent clustering methods predominantly utilize graph-based community detection approaches. Our proposition involves integrating biological knowledge as attributes of graph nodes to yield clustering outcomes that align more closely with biological interpretations. To realize this advancement, we have developed the MSGNN tool based on graph neural networks, enhancing the biological interpretation of complex data. |
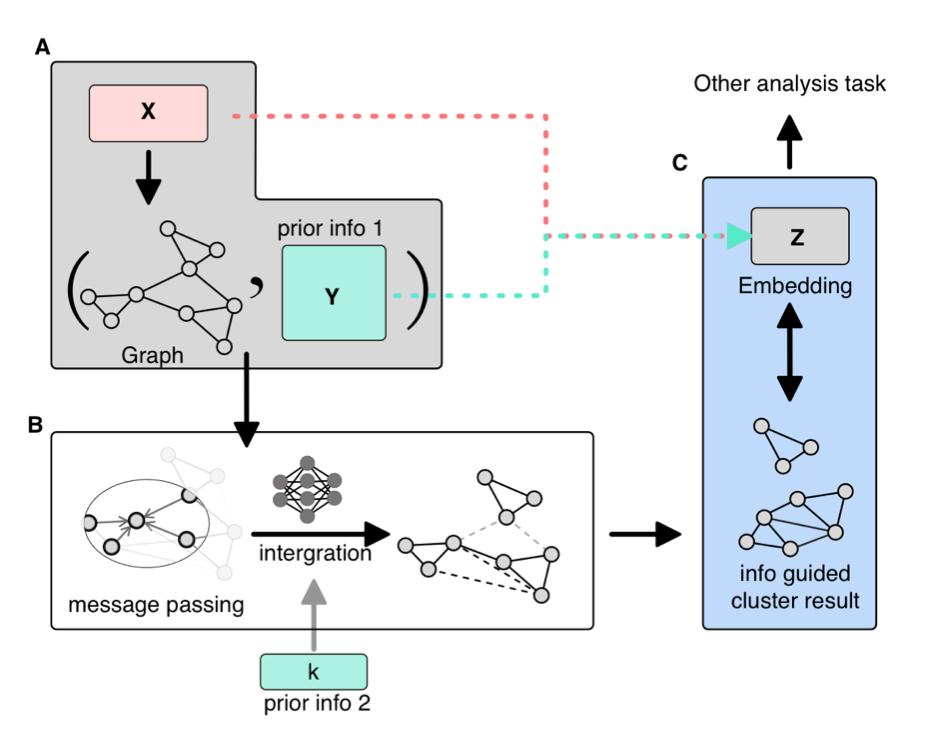
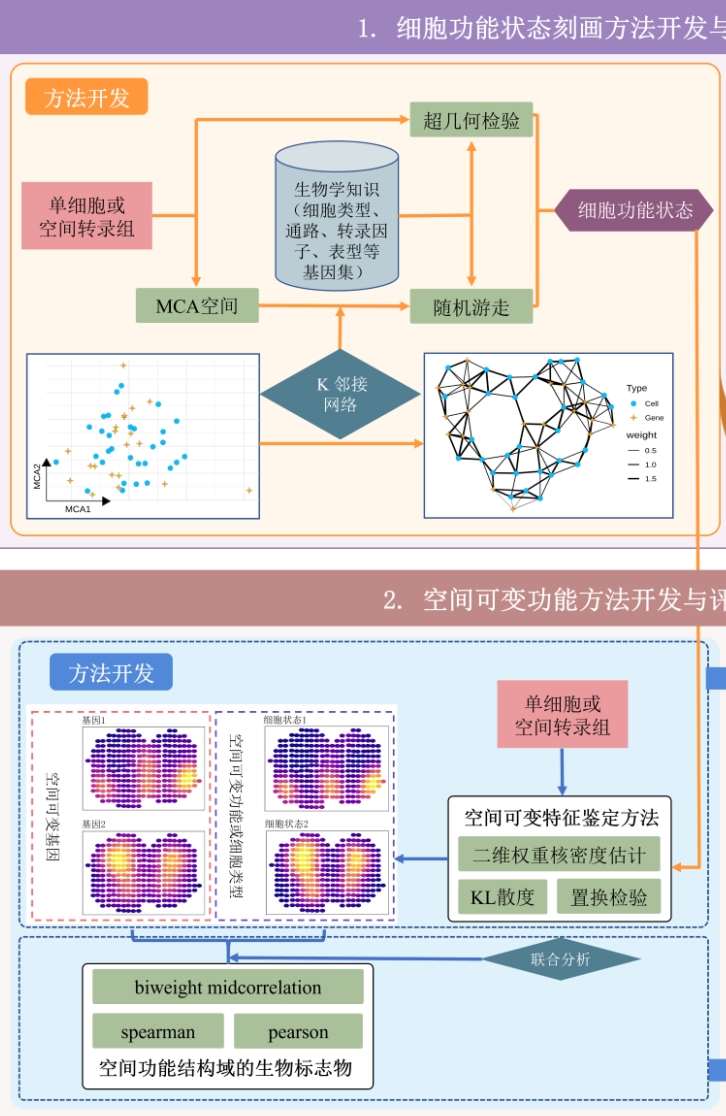
6. Identifying spatial variable biological functions Link to heading
|
|
Existing methods for identifying biological functions with spatially specific distributions typically begin with the identification of genes exhibiting high spatial variability. Subsequent enrichment analysis is then conductted to characterizing these functions. Our innovation lies in proposing a novel method for characterizing cell functional states, alongside a general approach for identifying spatially highly variable features. To operationalize these advancements, we have developed the SVP package. This tool facilitates the implementation of both methods, enabling the comprehensive characterizing various biological functions, including pathways, while also determining the spatial distribution specificity of these functions. |
{kind=link}
Feedback from the academic community Link to heading
|
Publications Link to heading
- G Yu*. Thirteen years of clusterProfiler. The Innovation. 2024, 5(6):100722.
- S Xu#, E Hu#, Y Cai#, Z Xie#, X Luo#, L Zhan, W Tang, Q Wang, B Liu, R Wang, W Xie, T Wu, L Xie, G Yu*. Using clusterProfiler to characterise Multi-Omics Data. Nature Protocols. 2024, 19(11):3292-3320.
- Q Wang#, M Li#, T Wu, L Zhan, L Li, M Chen, W Xie, Z Xie, E Hu, S Xu, G Yu*. Exploring epigenomic datasets by ChIPseeker. Current Protocols, 2022, 2(10): e585.
- N Sato, Y Tamada, G Yu, Y Okuno*. CBNplot: Bayesian network plots for enrichment analysis. Bioinformatics. 2022, 38(10):2959-2960.
- T Wu#, E Hu#, S Xu, M Chen, P Guo, Z Dai, T Feng, L Zhou, W Tang, L Zhan, X Fu, S Liu, X Bo*, G Yu*. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021, 2(3):100141.
- Yu G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. In: Kidder B. (eds) Stem Cell Transcriptional Networks. Methods in Molecular Biology, 2020, 2117:207-215. Humana, New York, NY.
- Z Hao, D Lv, Y Ge, J Shi, D Weijers, G Yu*, J Chen*. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Computer Science. 2020, 6:e251.
- G Yu. Using meshes for MeSH term enrichment and semantic analyses. Bioinformatics. 2018, 34(21):3766-3767.
- G Yu, QY He*. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Molecular BioSystems. 2016, 12(2):477-479.
- G Yu*, LG Wang, QY He*. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparision and visualization. Bioinformatics. 2015, 31(14):2382-2383.
- G Yu*, LG Wang, GR Yan, QY He*. DOSE: an R/Bioconductor package for Disease Ontology Semantic and Enrichment analysis. Bioinformatics. 2015, 31(4):608-609.
- G Yu, LG Wang, Y Han, QY He*. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.
- G Yu, QY He*. Functional similarity analysis of human virus-encoded miRNAs. Journal of Clinical Bioinformatics, 2011, 1(1):15.
- G Yu#, CL Xiao#, X Bo, CH Lu, Y Qin, S Zhan, QY He*. A new method for measuring functional similarity of microRNAs. Journal of Integrated OMICS, 2011, 1(1):49-54.
- G Yu#, F Li#, Y Qin, X Bo*, Y Wu, S Wang*. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010, 26(7):976-978.